Introduction
Liver transplantation is an effective treatment
strategy for a number of end-stage liver diseases including liver
failure, alcohol liver disease and liver cancer (1–3). Due
to the shortage of liver donors, transplantation from donors after
brain death (DBD) has increased. From 2010 to 2014, there were
2,897 cases of voluntary post-mortem donations in China (4), the majority of which were DBD.
However, compared with liver transplantation from a living donor,
the DBD liver transplant has more grafting complications, and acute
rejections of organs from DBD occur at a significantly higher rate
compared with the organs of living donors 6–24 months following
transplantation (5). The primary
causes of this are: Massive catecholamine release leading to
oxidative stress (6), release of
inflammatory mediators leading to systemic inflammatory response
(7,8) and pro-apoptotic caspase activation
(9,10). Brain death is independent of
hemodynamic stability, however, liver macro- and microcirculation
decreases, causing hepatic oxidative stress to increase and liver
cells to become ischemic-hypoxic (11). Oxidative stress has a major role in
organ transplant complications (12); however, the mechanism that induces
liver damage during brain death is not fully understood.
Peroxiredoxins are a widely distributed superfamily
of peroxidases that can eliminate reactive oxygen species (ROS)
(13). According to the number of
redox-active cysteines, peroxiredoxins can be classified as 1-Cys
or 2-Cys peroxiredoxin (Prdx). Prdx6 is a unique mammalian member
of the 1-Cys Prdx family (14).
Prdx6 is a bifunctional protein that exhibits peroxidase, as well
as phospholipase A2 (PLA2), activities (14). Prdx6 is expressed in the liver
(15) and protects cells from
damage by ROS induced by ischemia-reperfusion injury (16). By contrast, PLA2 activity is
important in the phospholipids of lung surfactant metabolism
(17), and is required for optimal
nicotinamide adenine dinucleotide phosphate-oxidase activity
(18). A recent study highlighted
that PLA2 activity has a substantial role in the protection of
cells against oxidative stress (19). Prdx6 is regulated via a number of
signaling pathways by nuclear factor-κB (NF-κB) (20), nuclear factor erythroid 2-related
factor 2 (21) and Jun N-terminal
kinase (22). However, to the best
of our knowledge, the function and mechanism of Prdx6 in liver
damage after brain death has not been reported in any previous
studies.
Therefore, we propose that Prdx6 may attenuate
ischemia and hypoxia-induced liver damage of DBD. In the present
study, the effects of Prdx6 on liver damage were investigated in
vivo and in vitro, and the underlying mechanisms were
explored. The present study determined that the expression of Prdx6
and inhibitor of κB-α (IκB-α) proteins are decreased in DBD liver
tissue. When normal liver cells (L02 cells) were exposed to
ischemia and hypoxia to mimic the conditions of brain death, cell
damage increased with exposure time. In addition, overexpression of
Prdx6 partially attenuated cell damage and the expression of Prdx6
appeared to be regulated by the NF-κB signaling pathway.
Furthermore, Prdx6 PLA2 activity has been revealed to be involved
in protecting cells against oxidative stress induced by ischemia
and hypoxia. Therefore, in agreement with our hypothesis, it is
considered that Prdx6 can attenuate liver damage after brain death.
These results provide valuable information to improve the quality
of donor livers and thus increase transplant success from DBD
livers.
Materials and methods
Cell culture and treatment
Normal human liver L02 cells were obtained from
Kunming Institute of Zoology (Chinese Academy of Sciences, Kunming,
China). The cells were grown to 70–80% confluence in 6-well culture
plates in HyClone™ Dulbecco's modified Eagle medium (DMEM; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml penicillin and 0.1 mg/ml streptomycin
(Beyotime Institute of Biotechnology, Nantong, China).
Liver damage following brain death was mimicked in
L02 cells by means of serum deprivation and hypoxia for 6, 12, 18
and 24 h. In order to simulate the ischemic and hypoxic
environment, L02 cells were cultured in HyClone serum-free DMEM/low
glucose medium (GE Healthcare Life Sciences), and exposed to a
gaseous mixture of 95% N2 and 5% CO2 (0%
O2) in a 37°C humidified incubator (Thermo Fisher
Scientific Inc.) for different time periods (0, 6, 12, 18, 24 h),
as previously described (23).
For the transfection experiment, L02 cells were
divided into three groups: A WT group (L02 cells cultured without
transfection), a vector group [L02 cells transfected with
pIRES2-ZsGreen1-vector plasmids (Wuhan Sanying Biotechnology, Inc.,
Wuhan, China)], and a Prdx6(+) group (L02 cells transfected with
pIRES2-ZsGreen1-Prdx6 plasmids). L02 cells were transfected with
pIRES2-ZsGreen1-Prdx6 plasmids using Lipofectamine 2000, according
to manufacturer's protocol (Invitrogen; Thermo Fisher Scientific,
Inc.), to over-express Prdx6. The cells were subjected to serum
deprivation and hypoxia for 12 h, then harvested for further
biochemical analyses 48 h after transfection. Transfection
efficiency was evaluated by green fluorescence levels as determined
by fluorescence microscopy, as well as protein expression
determined western blotting.
The activity of NF-κB was inhibited by pretreating
the cells with BAY11-7082 (5 µM; Santa Cruz Biotechnology
Inc., Dallas, TX, USA) for 1 h, prior to being exposed to the
ischemic-hypoxic conditions. The cells were then lysed using lysis
buffer (1 M Tris-HCl, pH 8.0, 5 M NaCl, and 1% NP-40) and harvested
for western blotting and CCK-8 assay.
To determine the protective effect of Prdx6 PLA2
against ischemia- and hypoxia-induced oxidative stress, L02 cells
(1×104) were treated with 0, 10, 20, 30 and 50 µM
MJ33, a PLA2 inhibitor. One group of cells was cultured with MJ33
for 0.5 h, then MJ33 was removed and the cells were cultured in
normal or ischemic-hypoxic conditions for 12 h (MJ33+0.5 h group).
The other group of cells was directly cultured with MJ33 in normal
or ischemic-hypoxic conditions for 12 h (MJ33+12 h group).
Determination of hepatocellular injury marker concentrations and
cell viability were then performed.
Clinical DBD samples, treatment and
ethical considerations
Liver tissue samples were collected from ten
patients with DBD and six patients that had accepted liver
hemangioma surgery, between February 2013 and December 2013 at the
Zhongnan Hospital of Wuhan University, Wuhan, China. The present
study was approved by the Ethics Committee of the Zhongnan Hospital
of Wuhan University and informed consent was obtained from all
patients prior to commencement of the study. Although the tissue
used for the control group was harvested from patients with liver
hemangioma it did not contain any cancerous tissue.
Western blot analysis
Cell lysate and liver tissue (from DBD and control
groups) samples were prepared in ice-cold radioimmunoprecipitation
assay lysis buffer (Applygen Technologies Inc., Beijing, China), as
previously described (24), then
centrifuged at 10,000 × g for 15 min at 4°C. The protein
concentration of the lysates was determined by a Bradford assay
(Beyotime Institute of Biotechnology). Equal quantities of protein
samples (30 µg) were loaded on a 15% sodium dodecyl sulfate
gel (60 V) and blotted onto polyvinylidene difluoride membrane (0.2
µm; EMD Millipore, Billerica, MA, USA). The membranes were
blocked with 5% skimmed milk at room temperature for 1 h. The
membranes were subsequently incubated at 4°C overnight with the
following specific primary antibodies: Rabbit polyclonal
anti-human/mouse/rat Prdx6 (1:500; cat. no. 13585-1-AP; Wuhan
Sanying Biotechnology, Inc.), mouse monoclonal anti-rat/human
p-IκB-α (1:200; cat. no. sc-8404; Santa Cruz Biotechnology, Inc.),
rabbit polyclonal anti-human/mouse/rat IκB-α (1:300; cat. no.
18220-1-AP; ProteinTech Group, Inc., Chicago, IL USA) and rabbit
polyclonal anti-human/mouse/rat β-actin (1:3,000; cat. no. BS1002;
Bioworld Technology Inc., St. Louis Park, MN, USA), then washed
three times with Tris-buffered saline and Tween-20 (TBST).
Subsequently, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit (1:3,000; cat. no. BL003A;
BioSharp, Hefei, China) or anti-mouse antibody (1:3,000; cat. no.
BL001A; BioSharp), and washed three times with TBST. The
immunoreactive proteins were visualized using a DAB Horseradish
Peroxidase Color Development enhanced chemiluminescence method kit
(cat. no. P0202; Beyotime Institute of Biotechnology).
Measurement of alanine transaminase
(ALT), aspartate transaminase (AST) and lactate dehydrogenase (LDH)
levels
ALT, AST and LDH were used as markers of
hepatocellular injury. L02 cells were exposed to ischemia and
hypoxia for different time periods (0, 6, 12, 18, 24 h).
Subsequently, ALT, AST and LDH concentration levels were measured
using an AU5400 Clinical Chemistry System (Beckman Coulter, Inc.,
Brea, CA, USA), which is an automatic biochemistry analyzer.
Measurement of intracellular ROS
The ROS in L02 cells were measured with a ROS assay
kit (Genmed Scientific Inc., Shanghai, China) according to
manufacturer's protocol. Subsequently, cells were cultured and
subjected to ischemia and hypoxia for different time periods. The
cells were maintained in the dark and incubated with working
solution (obtained from the ROS assay kit) at 37°C for 20 min;
resulting in the development of a green color proportional to the
amount of ROS present. Fluorescence was monitored by
excitation/emission of 490/530 nm using a fluorescence microscope
(IX71-A12FL; Olympus Corporation, Tokyo, Japan), and fluorescence
intensity of 10,000 cells was measured using an FC500 flow
cytometer (Beckman Coulter, Inc.).
Cell Counting Kit-8 (CCK-8) viability
assay
Cell viability was assessed using a CCK-8 detection
kit (Dojindo Molecular Technologies, Inc., Kumamoto, Japan),
according to the manufacturer's protocol. Briefly, 1×105
cells/well were cultured in 96-well plates. Cells were treated with
BAY11-7082 or MJ33, as described, and exposed to ischemic and
hypoxic conditions for different periods of time. Subsequently, 100
µl 10% CCK-8 solution was added per well and then incubated
at 37°C for 1 h. Absorbance was measured at 450 nm using a
microplate reader (SpectraMax 190; Eppendorf, Hamburg,
Germany).
Measurement of PLA2 enzymatic
activity
PLA2 activity was measured using a PLA2 Assay kit
(Invitrogen, Thermo Fisher Scientific Inc.), according to the
manufacturer's instructions. Fluorescence was measured using a
microplate reader (SpectraMax 190; Molecular Devices, LLC,
Sunnyvale, CA, USA) with excitation and emission wavelengths of 485
and 520 nm, respectively. The effect of
1-hexadecyl-3-trifluoroethylglycero-sn-2-phosphomethanol (MJ33;
Sigma-Aldrich, St. Louis, MO, USA) on PLA2 activity was measured.
Three MJ33 treatments were applied to the L02 cells: i) Following a
0.5 h treatment with MJ33 PLA2 enzymatic activity in L02 cells was
measured immediately; ii) Following a 0.5 h treatment with MJ33
PLA2 enzymatic activity in L02 cells was measured after 12 h; and
iii) Following a 12 h treatment with MJ33, PLA2 enzymatic activity
in L02 cells was measured immediately. The concentrations of MJ33
used were 10, 20, 30 and 50 µM.
Statistical analysis
The results are expressed as the mean ± standard
deviation for three or more independent experiments. One-way
analysis of variance and independent Student's t-test were
performed using SPSS software (version 11.5; SPSS, Inc., Chicago,
IL, USA). P<0.05 indicated a statistically significant
difference.
Results
Prdx6 expression is decreased and the
NF-κB signaling pathway is activated in DBD livers
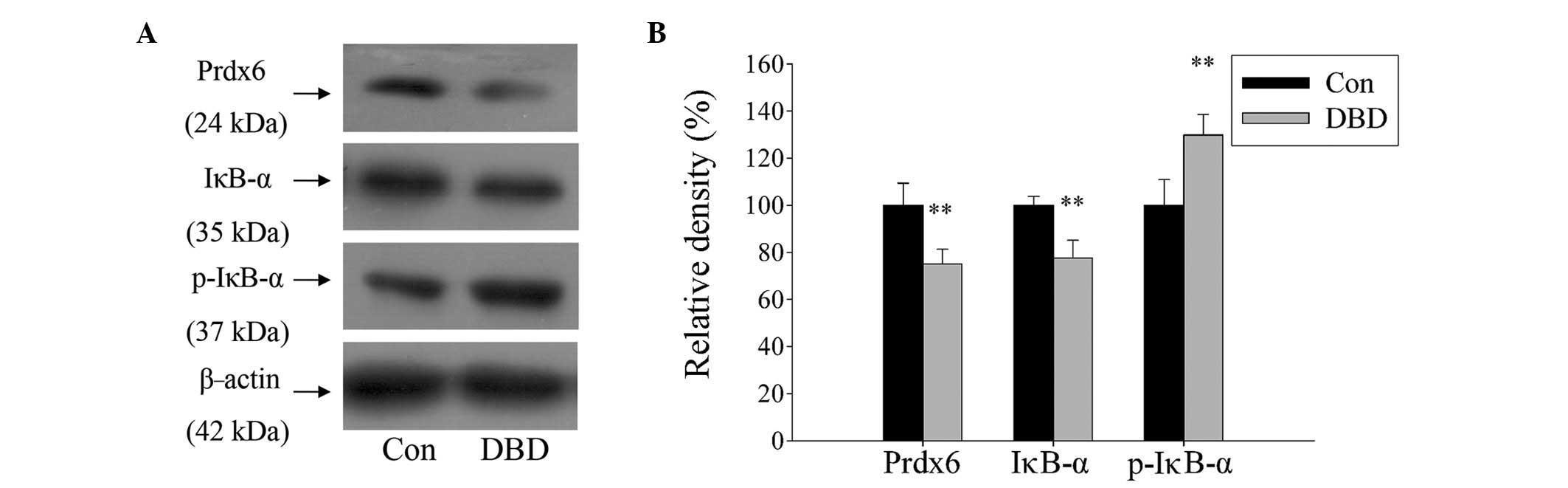
The role of Prdx6 and its regulation by NF-κB in DBD
livers was explored. A total of ten DBD patients and six normal
controls were examined. The present study identified that the
expression of Prdx6 in liver tissue was significantly reduced in
the DBD group compared with the control group (P<0.01).
Simultaneously, the expression of IκB-α was significantly reduced
and its phosphorylated form (p-IκB-α) was significantly increased
in DBD (Fig. 1; P>0.01). The
results suggest that Prdx6 may be involved in liver damage
processes and that its expression is regulated by the NF-κB
signaling pathway.
Prdx6 may attenuate ischemic and hypoxic
damage in L02 cells
In order to determine the role of Prdx6 in DBD
livers, L02 cells were cultured under ischemic-hypoxic conditions
to mimic the state of brain death. AST and LDH concentration levels
were demonstrated to significantly increase (Fig. 2A–C) and cell viability was
significantly reduced as cell exposure time to DBD conditions was
increased (Fig. 2D). However, the
increase in levels of ALT were not as marked as those of AST, which
suggests that ALT may be located in the cytoplasm, and AST in the
mitochondria. ALT, AST and LDH concentration levels were identified
to significantly increase (Fig.
2A–C) and cell viability was significantly reduced as cell
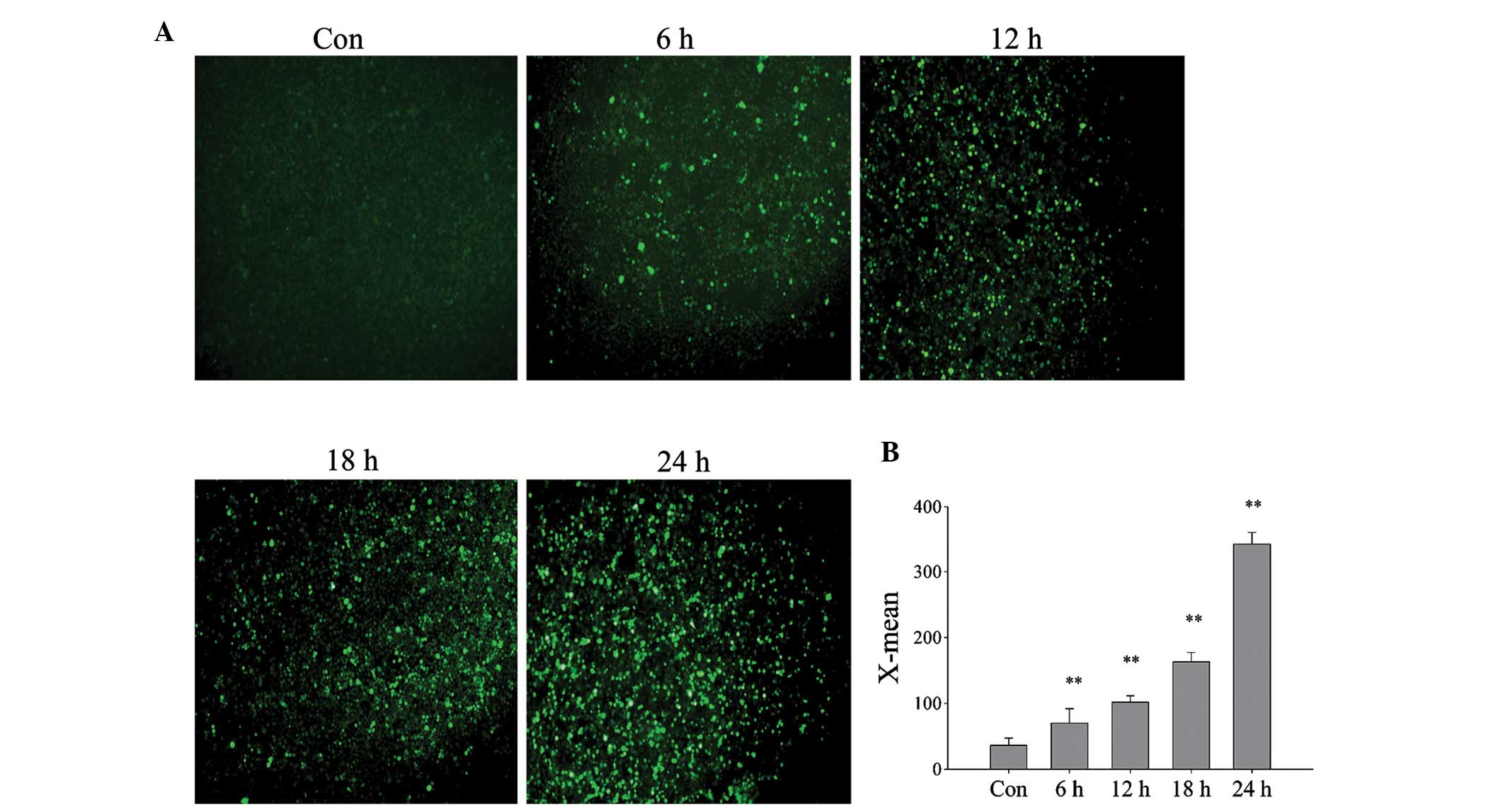
exposure time to DBD conditions was increased (Fig. 2D). Using an ROS assay kit and a
fluorescence microscope, it was detected that the intracellular ROS
levels of L02 cells also increase with exposure time (Fig. 3A), and the relative fluorescence
value significantly increased (Fig.
3B; P<0.01). This suggests that cell damage occurs in a
time-dependent manner that may be caused by increased ROS.
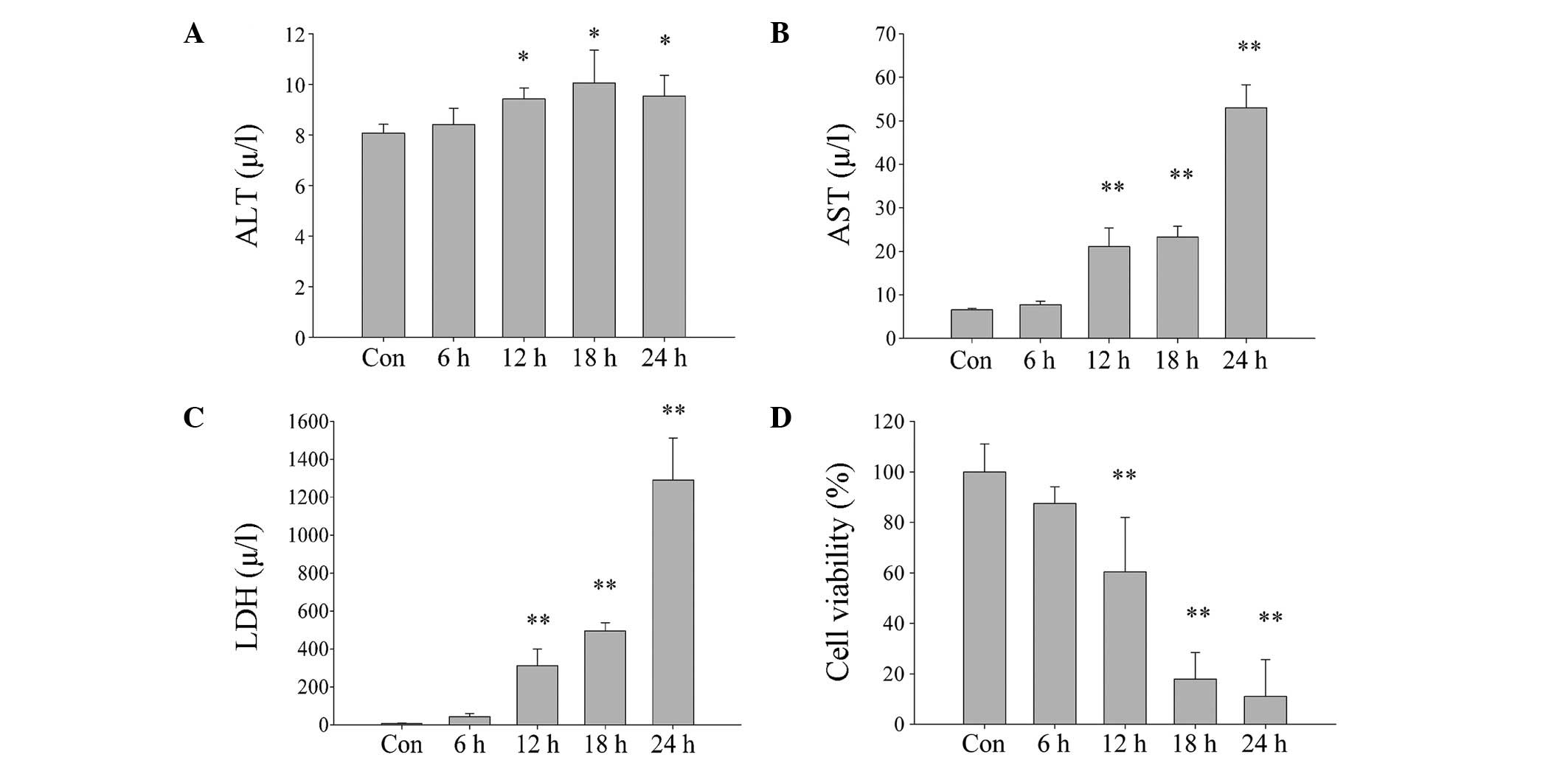 | Figure 2Damage of L02 cells exposed to
ischemia and hypoxia increases with increased exposure times (0, 6,
12, 18 and 24 h). (A) ALT, (B) AST and (C) LDH concentration levels
measured in cell culture following exposure to ischemia and
hypoxia. (D) Survival rate of cells with increased exposure to
hypoxia and ischemia, as determined by Cell Counting Kit-8 assay.
Data are expressed as mean ± standard deviation
*P<0.05, **P<0.01 vs. Con. ALT, alanine
transaminase; Con, control; AST, aspartate transaminase; LDH,
lactate dehydrogenase. |
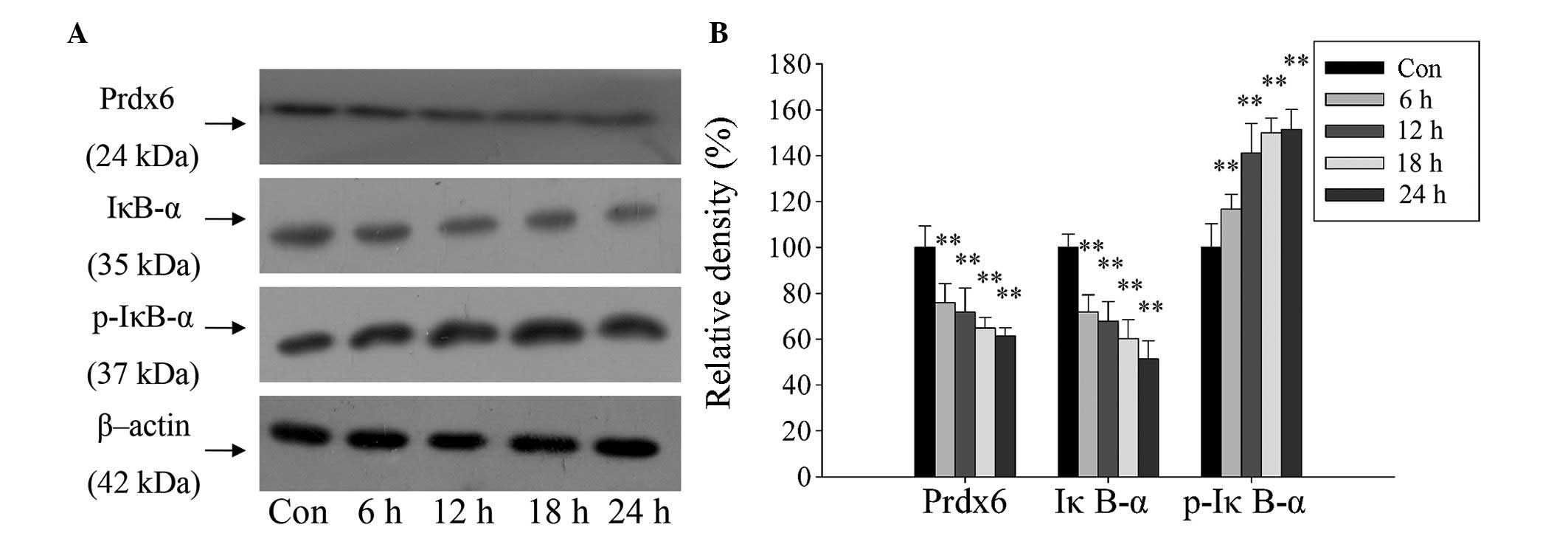
Furthermore, a western blot assay revealed the same
expression pattern for Prdx6, IκB-α and p-IκB-α as in the DBD liver
samples. The expression of Prdx6 and IκB-α proteins in L02 cells
were significantly reduced, and the expression of p-IκB-α protein
significantly increased over time (Fig. 4; P<0.01). The changes in
concentration of IκB-α and p-IκB-α indicate that the NF-κB
signaling pathway was activated. This demonstrates the potential
stress-preventive role of Prdx6 in L02 cells under ischemia and
hypoxia exposure, and indicates that Prdx6 may be regulated by
NF-κB.
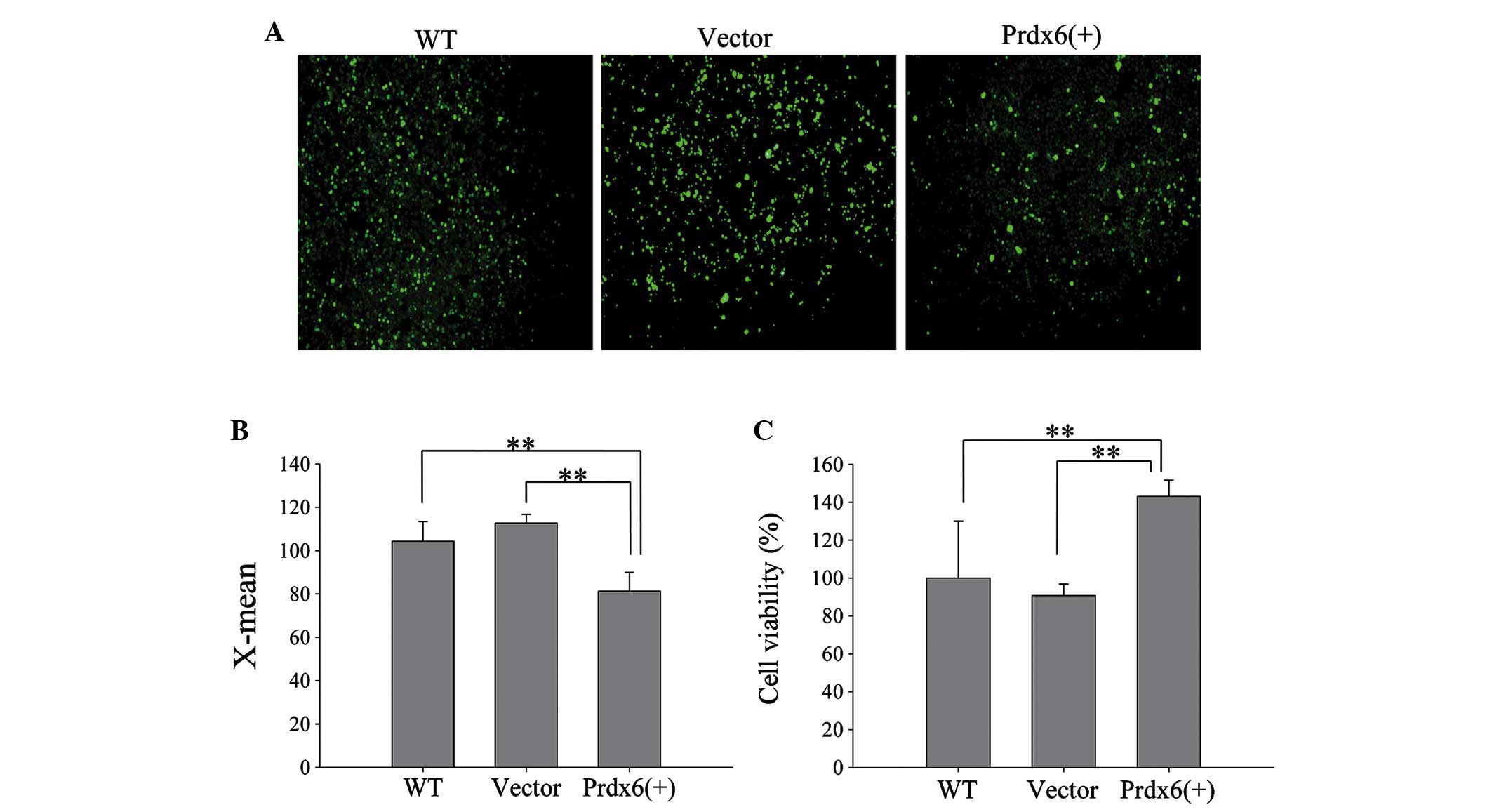
The efficiency of L 02 transfection wit h
pIRES2-2sGreen1-Prdx6 was evaluated using green fluorescence
(Fig. 5A) and western blotting
(Fig. 5B). L02 cells significantly
overexpressed Prdx6 compared with the controls (Fig. 5C; P<0.01). Furthermore, L02
cells overexpressing Prdx6 exhibited significantly reduced levels
of intracellular ROS when exposed to ischemia and hypoxia (Fig. 6A and B; P<0.01), and cell
viability significantly increased in these cells versus the control
cells (Fig. 6C; P<0.01). The
results confirm that Prdx6 has a protective function against cell
death caused by ischemia and hypoxia-induced oxidative stress by
optimizing ROS levels.
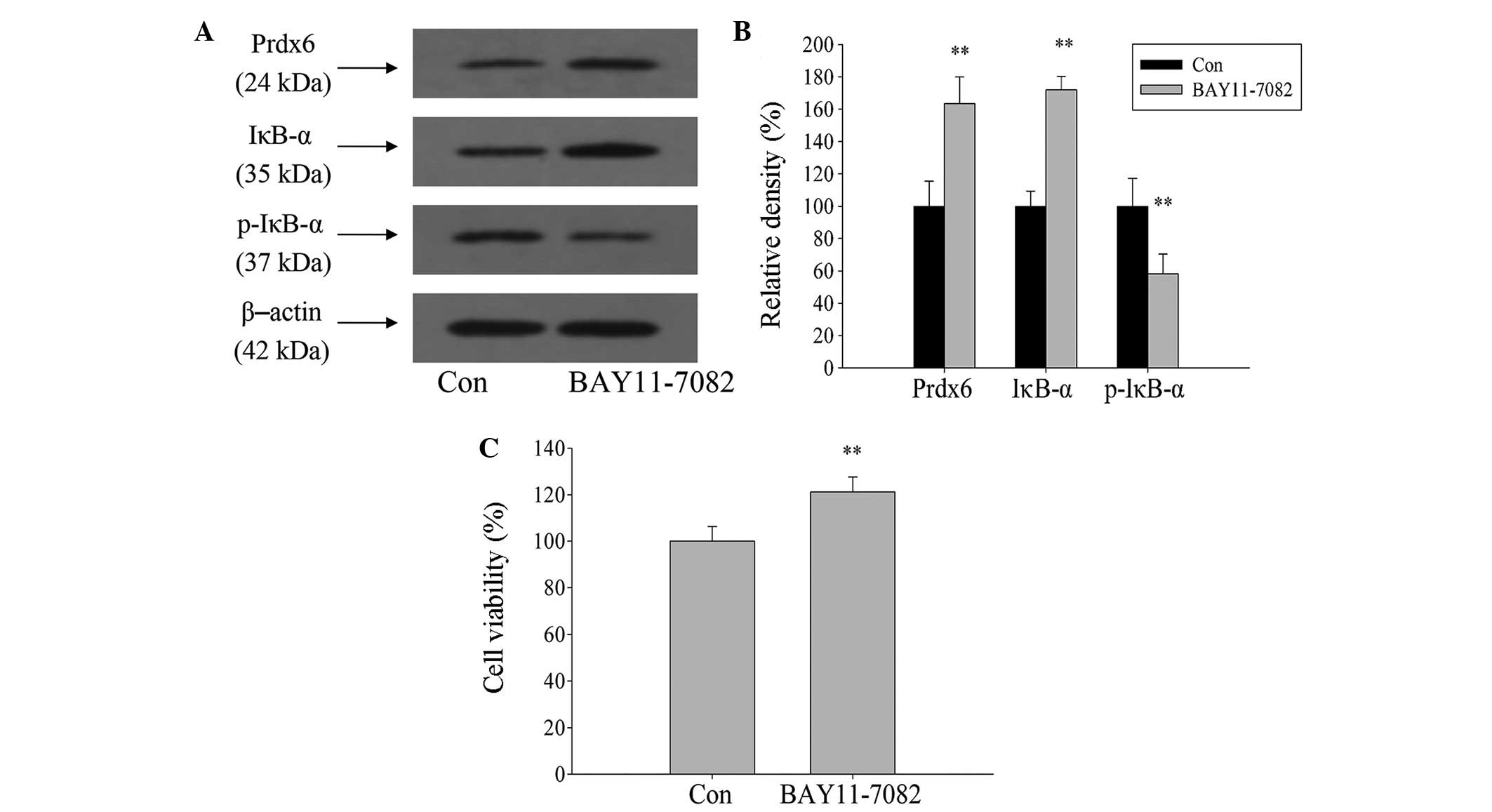
NF-κB signaling pathway negatively
regulates Prdx6 expression
NF-κB was activated during ischemia and hypoxia. To
clarify whether NF-κB activation is involved in the regulation of
Prdx6 expression in L02 cells during ischemia and hypoxia,
BAY11-7082 was used to inhibit the activation of NF-κB. Western
blot analysis showed that expression levels of IκB-α and Prdx6
significantly increased, and p-IκB-α expression significantly
decreased after 1 h treatment of L02 cells with BAY11-7082, when
compared with the control (Fig. 7A and
B; P<0.01). Following a 12-h exposure to ischemia and
hypoxia, cell viability significantly increased compared with the
control group (Fig. 7C;
P<0.01). Therefore, this indicates that Prdx6 expression is
negatively regulated by NF-κB during ischemia and hypoxia.
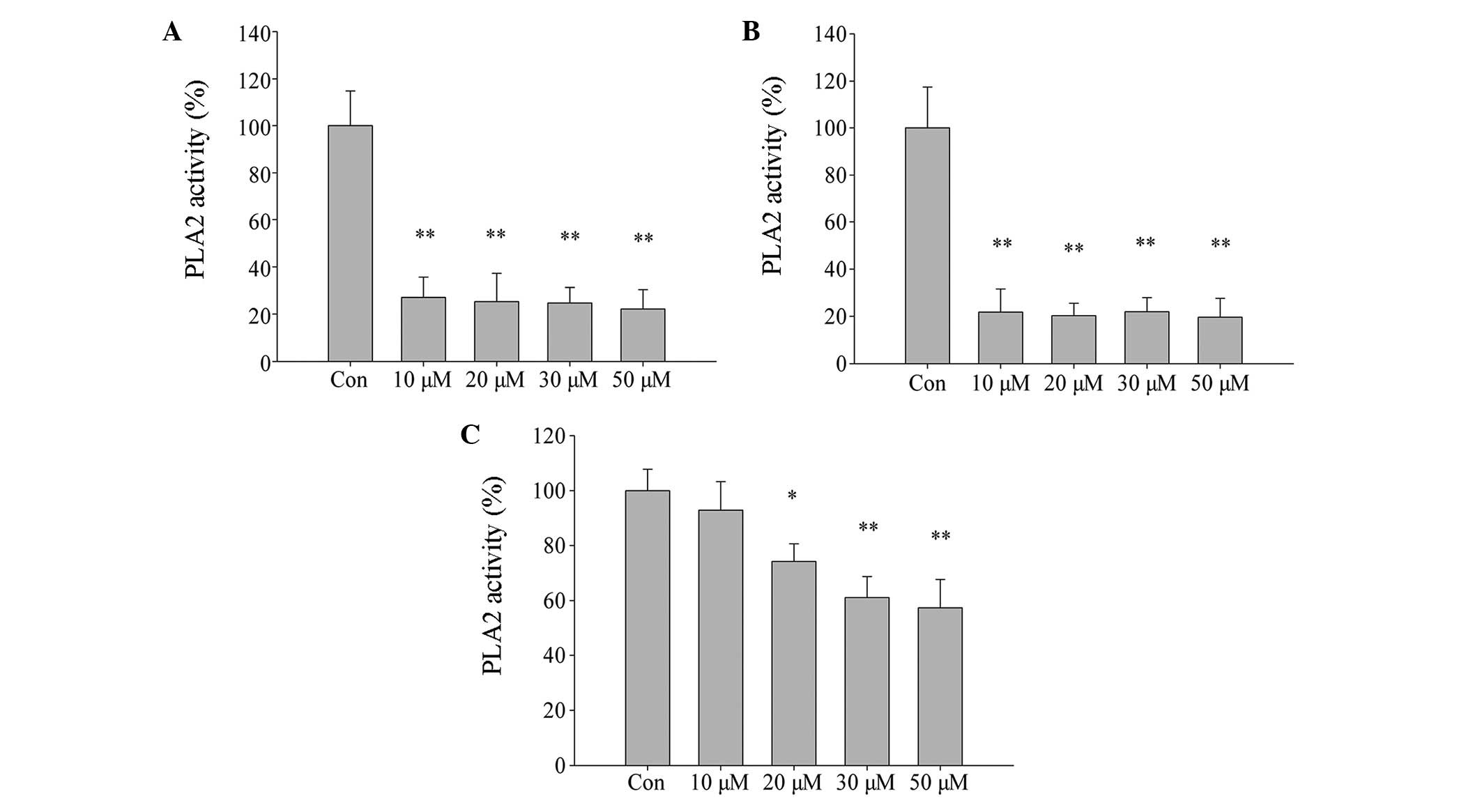
Inhibition of PLA2 activity by MJ33
exacerbates ischemia and hypoxia-induced cell injury in L02
cells
The role of Prdx6 PLA2 in oxidative stress induced
by ischemia and hypoxia was explored. Initially, L02 cells were
treated with different concentrations of MJ33 for two different
time periods (MJ33+0.5 h and MJ33+12 h), without exposure to
ischemia-hypoxia. There was no significant difference in cell
viability between the two MJ33 treatment groups (Fig. 8A), thus cytotoxicity of MJ33 at
concentrations of <50 µM was not evident. However, when
the cells were pretreated with MJ33 and then exposed to ischemic
and hypoxic conditions (MJ33+0.5 h) or concurrently treated with
MJ33 and ischemia-hypoxia (MJ33+12 h), a significant difference in
cell viability was observed. The cell viability in each group
declined as MJ33 concentration increased. Notably, the cell
viability of the MJ33+0.5 h group was marginally higher than that
of the MJ33+12 h group cells (Fig.
8B). The ALT concentration level in cells exposed to culture
medium containing 0 µM (control) and 20 µM MJ33
exhibited no significant difference, while AST and LDH levels were
significantly increased in both MJ33 treatment groups. However,
those in the MJ33+0.5 h group exhibited lower concentrations than
MJ33+12 h group cells, which coincides with the CCK-8 data
(Fig. 8C).
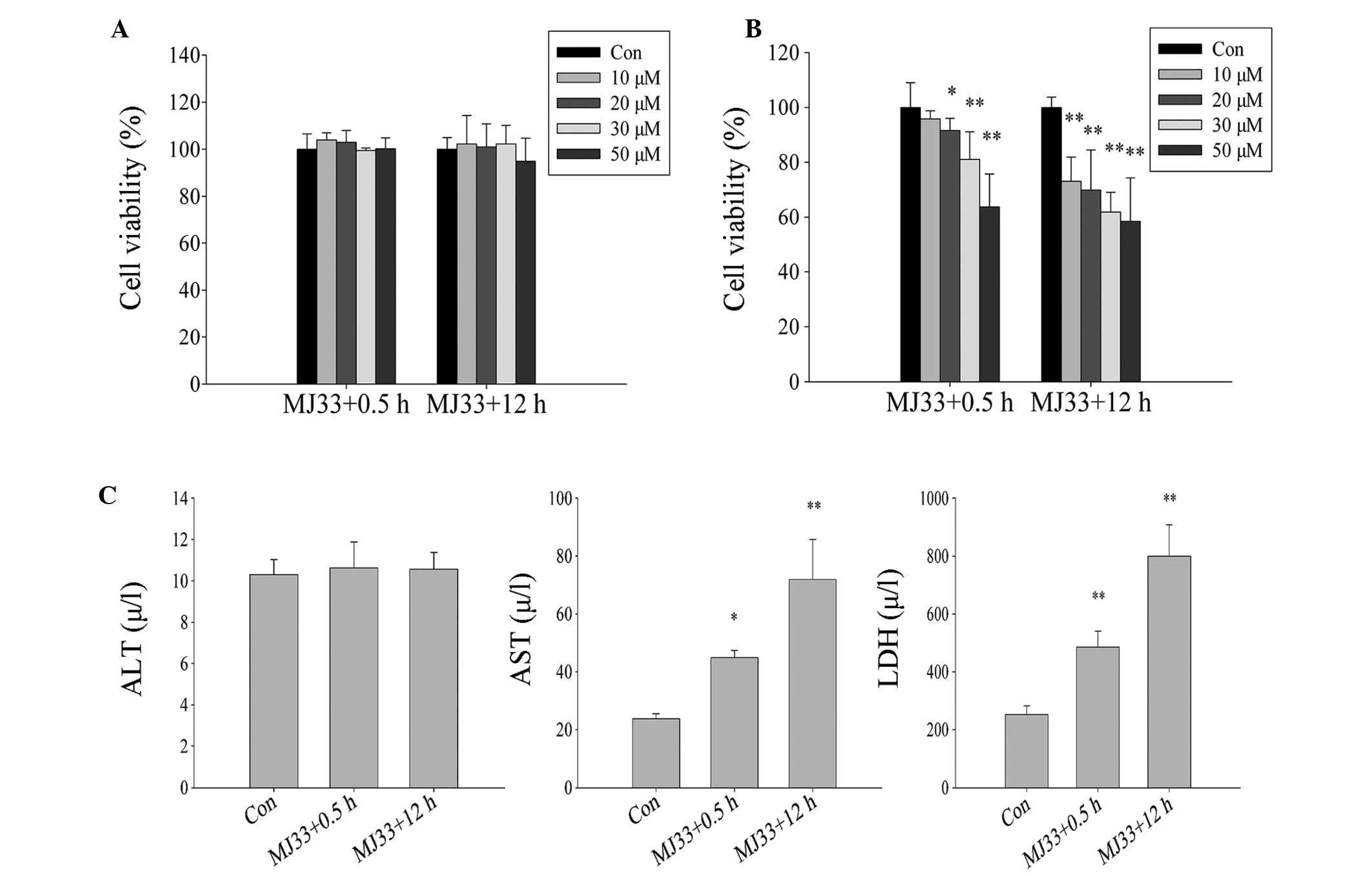 | Figure 8Effect of inhibition of PLA2 activity
by MJ33 (0, 10, 20, 30 and 50 µM) on L02 cells damage when
subjected to ischemia and hypoxia. Viability of cells following (A)
treatment with MJ33 and following (B) treatment with MJ33 and
exposure to ischemic and hypoxic conditions. (C) The damage L02
cells suffered following exposure to ischemia and hypoxia was
measured by ALT, AST and LDH concentration levels. MJ33+0.5 h
group, cultured with MJ33 for 0.5 h, then MJ33 was removed and the
cells were cultured in normal or ischemic-hypoxic conditions for 12
h; MJ33+12 h group, directly cultured with MJ33 in normal or
ischemic-hypoxic conditions for 12 h. *P<0.05,
**P<0.01 vs. control. ALT, alanine transaminase; Con,
control; AST, aspartate transaminase; LDH, lactate
dehydrogenase. |
The effect of MJ33 on cell PLA2 activity was
evaluated using an assay kit. PLA2 activity was significantly
inhibited by treatment of L02 cells with increasing concentrations
of MJ33 for 0.5 h and 12 h, then immediately harvested (Fig. 9A and B, respectively; P<0.01).
However, in cells that were pretreated with MJ33 for 0.5 h and then
cultured for 12 h after MJ33 removal, PLA2 activity did not
decrease as significantly compared with the other two groups
(Fig. 9C). This may explain why
cell viability was higher in the equivalent MJ33+0.5 h group of
cells. Thus, the current results indicate that the Prdx6 PLA2
activity is involved in cell protection against oxidative stress
caused by ischemia and hypoxia.
Discussion
The present study identified a reduction in the
expression of Prdx6 in DBD livers. Liver cells from DBD are
considered to be subjected to ischemic-hypoxic damage, and Prdx6 is
an antioxidant protein that can protect cells from oxidative stress
(15). Thus, Prdx6 is possibly
involved in the prevention of ischemia- and hypoxia-induced liver
damage.
L02 cells were subjected to ischemia and hypoxia to
mimic DBD liver cells. The present study determined that the
expression of Prdx6 was reduced in L02 cells. Furthermore, ALT, AST
and LDH concentration levels were increased, in addition to
intracellular ROS levels, resulting in reduced cell viability. In a
previous study, Prdx6 expression was downregulated upon serum
deprivation in mouse liver cells (25). In addition, it has been reported
that retinal ganglion cells exposed to hypoxia demonstrate reduced
expression of Prdx6 with higher ROS levels and increased cell death
(26). The current findings are
consistent with these studies, as it was observed that
over-expression of Prdx6 could reduce the levels of intracellular
ROS and improve cell viability. ROS can activate apoptosis in
hepatocytes (27), therefore, the
increased cell damage possibly results from higher levels of
intracellular ROS, which are in turn caused by the reduced
expression of Prdx6, indicating that Prdx6 has a protective role
when liver cells are exposed to ischemia and hypoxia.
In the present study, the expression of IκB-α was
reduced and p-IκB-α was increased in DBD liver tissue and
ischemic-hypoxic L02 cells, indicating that NF-κB activity was
increased. However, there remain inconsistencies in the association
between Prdx6 and NF-κB. For example, previous studies have
reported that upregulation of Prdx6 may inhibit the activation of
NF-κB (26,28,29).
Other studies state that NF-κB may negatively regulate Prdx6
expression in oxidative stress, as observed in mice livers treated
with ethanol (30) and mouse
hippocampal cells subjected to hypoxia (20). BAY11-7082, a specific IκB
kinase/NF-κB inhibitor, was used to inhibit NF-κB transcriptional
activity in L02 cells in the present study, resulting in increased
expression of Prdx6 protein and cell viability when cells were
exposed to ischemia and hypoxia. Previous studies revealed that ROS
could activate NF-κB in cell-type specific molecular mechanisms
(31,32). Therefore, it is possible that NF-κB
is an important signaling pathway that mediates Prdx6 expression in
DBD ischemic-hypoxic liver cells, leading to elevated ROS
activating NF-κB and resulting in the inhibition of Prdx6
expression. This, in turn, leads to ROS elevation, triggering a
positive-feedback mechanism and causing more liver injury.
To the best of our knowledge, the present study
observed for the first time, that Prdx6 PLA2 activity has a
protective role in liver cells. ROS can peroxidase unsaturated
fatty acids in the phospholipids of cellular membranes, thus
causing impaired membrane function (33). PLA2 can hydrolyze an acyl or alkyl
linkage at the sn-2 position of phospholipids to produce free fatty
acids (34). Therefore, PLA2
contributes to membrane repair by releasing free fatty acids and
forming 2-lysophospho-lipid acceptors that may be reacylated by
acyltransferases (35). Although
Prdx6 PLA2 activity is high at pH 4 and low at pH 7, as Prdx6 binds
to oxidize phospholipids at pH 7, PLA2 activity increases (36). PLA2 activity is not as efficient in
the reduction of peroxidized phospholipids as Prdx6 peroxidase
activity, however it is still an important alternative pathway in
the event of a decline in peroxidase activity due to reduced Prdx6
expression.
In conclusion, the current study observed that the
Prdx6 expression was reduced in DBD liver tissue, and liver cells
subjected to ischemia and hypoxia. This was associated with liver
injury. Conversely, overexpression of Prdx6 was found to partially
reverse the liver cell damage. In addition, expression of Prdx6
appeared to be regulated by the NF-κB signaling pathway, and Prdx6
PLA2 activity contributed to cell protection during ischemia and
hypoxia. Thus, the present study provides a novel perspective on
the protection mechanism against liver damage after brain death,
and provides valuable information for the development of a novel
strategy for the improvement of donor liver quality.
References
|
1
|
Telles-Correia D and Mega I: Candidates
for liver transplantation with alcoholic liver disease:
Psychosocial aspects. World J Gastroenterol. 21:11027–11033. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heimbach JK, Hirose R, Stock PG, Schladt
DP, Xiong H, Liu J, Olthoff KM, Harper A, Snyder JJ, Israni AK, et
al: Delayed hepatocellular carcinoma model for end-stage liver
disease exception score improves disparity in access to liver
transplant in the United States. Hepatology. 61:1643–1650. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park SJ, Lim YS, Hwang S, Heo NY, Lee HC,
Suh DJ, Yu E and Lee SG: Emergency adult-to-adult living-donor
liver transplantation for acute liver failure in a hepatitis B
virus endemic area. Hepatology. 51:903–911. 2010.
|
|
4
|
Huang JF, Wang HB, Zheng SS, Liu YF, Shi
BY, Shen ZY, Hu SS, Ye QF, Xue WJ and He XS: Advances in China's
organ transplantation achieved with the guidance of law. Chin Med J
(Engl). 128:143–146. 2015. View Article : Google Scholar
|
|
5
|
Weiss S, Kotsch K, Francuski M,
Reutzel-Selke A, Mantouvalou L, Klemz R, Kuecuek O, Jonas S,
Wesslau C, Ulrich F, et al: Brain death activates donor organs and
is associated with a worse I/R injury after liver transplantation.
Am J Transplant. 7:1584–1593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barklin A, Larsson A, Vestergaard C,
Koefoed-Nielsen J, Bach A, Nyboe R, Wogensen L and Tønnesen E: Does
brain death induce a pro-inflammatory response at the organ level
in a porcine model? Acta Anaesthesiol Scand. 52:621–627. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kusaka M, Pratschke J, Wilhelm MJ, Ziai F,
Zandi-Nejad K, Mackenzie HS, Hancock WW and Tilney NL: Activation
of inflammatory mediators in rat renal isografts by donor brain
death. Transplantation. 69:405–410. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takada M, Nadeau KC, Hancock WW, Mackenzie
HS, Shaw GD, Waaga AM, Chandraker A, Sayegh MH and Tilney NL:
Effects of explosive brain death on cytokine activation of
peripheral organs in the rat. Transplantation. 5:1533–1542. 1998.
View Article : Google Scholar
|
|
9
|
Van Der Hoeven JA, Moshage H, Schuurs T,
Nijboer M, Van Schilfgaarde R and Ploeg RJ: Brain death induces
apoptosis in donor liver of the rat. Transplantation. 76:1150–1154.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adrie C, Monchi M, Fulgencio JP, Cottias
P, Haouache H, Alvarez-Gonzalvez A, Guerrini P, Cavaillon JM and
Adib-Conquy M: Immune status and apoptosis activation during brain
death. Shock. 33:353–362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Golling M, Mehrabi A, Blum K, Jahnke C,
Kellner H, Bud O, Hashemi B, Breitkreutz R, Becker-Brandenbutg K,
Schemmer P, et al: Effects of hemodynamic instability on brain
death-induced prepreservation liver damage. Transplantation.
75:1154–1159. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dutkiewicz G, Domanski L, Binczak-Kuleta
A, Pawlik A, Safranow K, Dziedziejko V, Wisniewska M, Ciechanowicz
A and Ciechanowski K: Lack of association of polymorphisms
239+34A/C in the SOD1 gene and 47C/T in the SOD2 gene with delayed
graft function and acute and chronic rejection of kidney
allografts. Transplant Proc. 41:3701–3703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rhee SG, Kang SW, Chang TS, Jeong W and
Kim K: Peroxiredoxin, a novel family of peroxidases. IUBMB Life.
52:35–41. 2001. View Article : Google Scholar
|
|
14
|
Fisher AB: Peroxiredoxin 6: A bifunctional
enzyme with glutathione peroxidase and phospholipase A2
activities. Antioxid Redox Signal. 15:831–844. 2011. View Article : Google Scholar :
|
|
15
|
Simeone M and Phelan SA: Transcripts
associated with Prdx6 (peroxiredoxin 6) and related genes in mouse.
Mamm Genome. 16:103–111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eismann T, Huber N Shin T, Kuboki S,
Galloway E, Wyder M, Edwards MJ, Greis KD, Shertzer HG, Fisher AB
and Lentsch AB: Peroxiredoxin-6 protects against mitochondrial
dysfunction and liver injury during ischemia-reperfusion in mice.
Am J Physiol Gastrointest Liver Physiol. 296:G266–G274. 2009.
View Article : Google Scholar :
|
|
17
|
Fisher AB, Dodia C, Feinstein SI and Ho
YS: Altered lung phospholipid metabolism in mice with targeted
deletion of lysosomal-type phospholipase A2. J Lipid Res.
46:1248–1256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ambruso DR, Ellison MA, Thurman GW and
Leto TL: Peroxiredoxin 6 translocates to the plasma membrane during
neutrophil activation and is required for optimal NADPH oxidase
activity. Biochim Biophys Acta. 1823.306–315. 2012.
|
|
19
|
Lien YC, Feinstein SI, Dodia C and Fisher
AB: The roles of peroxidase and phospholipase A2 activities of
peroxiredoxin 6 in protecting pulmonary microvascular endothelial
cells against peroxidative stress. Antioxid Redox Signal.
16:440–451. 2012. View Article : Google Scholar :
|
|
20
|
Chhunchha B, Fatma N, Kubo E, Rai P, Singh
SP and Singh DP: Curcumin abates hypoxia-induced oxidative stress
based-ER stress-mediated cell death in mouse hippocampal cells
(HT22) by controlling Prdx6 and NF-κB regulation. Am J Physiol Cell
Physiol. 304:C636–C655. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chowdhury I, Fisher AB,
Christofidou-Solomidou M, Gao L, Tao JQ, Sorokina EM, Lien YC,
Bates SR and Feinstein SI: Keratinocyte growth factor and
glucocorticoid induction of human peroxiredoxin 6 gene expression
occur by independent mechanisms that are synergistic. Antioxid
Redox Signal. 20:391–402. 2014. View Article : Google Scholar :
|
|
22
|
Paula FM, Ferreira SM, Boschero AC and
Souza KL: Modulation of the peroxiredoxin system by cytokines in
insulin-producing RINm5F cells: Down-regulation of PRDX6 increases
susceptibility of beta cells to oxidative stress. Mol Cell
Endocrinol. 374:56–64. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen K, Li YH, Xu SQ, Hu SH and Zhang L:
Protective effects of peroxisome proliferator-activated receptor-α
agonist, Wy14643, on hypoxia/reoxygenation injury in primary rat
hepatocytes. PPAR Res. 2012:5479802012. View Article : Google Scholar
|
|
24
|
Chhunchha B, Fatma N, Bhargavan B, Kubo E,
Kumar A and Singh DP: Specificity protein, Sp1-mediated increased
expression of Prdx6 as a curcumin-induced antioxidant defense in
lens epithelial cells against oxidative stress. Cell Death Dis.
2:e2342011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gallagher BM and Phelan SA: Investigating
transcriptional regulation of Prdx6 in mouse liver cells. Free
Radic Biol Med. 42:1270–1277. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tulsawani R, Kelly LS, Fatma N, Chhunchha
B, Kubo E, Kumar A and Singh DP: Neuroprotective effect of
peroxiredoxin 6 against hypoxia-induced retinal ganglion cell
damage. BMC Neurosci. 11:1252010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Xu Y, Wang H, Xue P, Li X, Li B,
Zheng Q and Sun G: Arsenic induces mitochondria-dependent apoptosis
by reactive oxygen species generation rather than glutathione
depletion in Chang human hepatocytes. Arch Toxicol. 83:899–908.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fatma N, Kubo E, Sen M, Agarwal N,
Thoreson WB, Camras CB and Singh DP: Peroxiredoxin 6 delivery
attenuates TNF-alpha-and glutamate-induced retinal ganglion cell
death by limiting ROS levels and maintaining Ca2+
homeostasis. Brain Res. 1233:63–78. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun XG, Fu XQ, Cai HB, Liu Q, Li CH, Liu
YW, Li YJ, Liu ZF, Song YH and Lv ZP: Proteomic analysis of
protective effects of polysaccharides from Salvia miltiorrhiza
against immunological liver injury in mice. Phytother Res.
25:1087–1094. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roede JR, Stewart BJ and Petersen DR:
Decreased expression of peroxiredoxin 6 in a mouse model of ethanol
consumption. Free Radic Biol Med. 45:1551–1558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gloire G, Legrand-Poels S and Piette J:
NF-kappaB activation by reactive oxygen species: Fifteen years
later. Biochem Pharmacol. 72:1493–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang H, Wang ZW, Wu HB, Li Z, Li LC, Hu
XP, Ren ZL, Li BJ and Hu ZP: Transforming growth factor-β1 induces
matrix metal-loproteinase-9 expression in rat vascular smooth
muscle cells via ROS-dependent ERK-NF-κB pathways. Mol Cell
Biochem. 375:11–21. 2013.PubMed/NCBI
|
|
33
|
Girotti AW: Lipid hydroperoxide
generation, turnover, and effector action in biological systems. J
Lipid Res. 39:1529–1542. 1998.PubMed/NCBI
|
|
34
|
Schaloske RH and Dennis EA: The
phospholipase A2 superfamily and its group numbering system.
Biochim Biophys Acta. 1761:1246–1259. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Winstead MV, Balsinde J and Dennis EA:
Calcium-independent phospholipase A(2): Structure and function.
Biochim Biophys Acta. 1488:28–39. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Manevich Y, Shuvaeva T, Dodia C, Kazi A,
Feinstein SI and Fisher AB: Binding of peroxiredoxin 6 to substrate
determines differential phospholipid hydroperoxide peroxidase and
phospholipase A(2) activities. Arch Biochem Biophys. 485:139–149.
2009. View Article : Google Scholar : PubMed/NCBI
|