Introduction
Colorectal cancer is a major cause of
cancer-associated mortality worldwide (1). Although colorectal cancer can be
effectively managed if detected early, the prognosis of patients
with colorectal cancer is poor due to the recurrence and distant
metastasis (2). It is therefore of
high importance to identify proteins involved in colorectal
tumorigenesis, which may be utilized as targets for therapeutic or
diagnostic strategies.
Tumorigenesis is closely associated with genetic
alterations and epigenetic modifications (3). Epigenetic modification results from
promoter methylation and/or alterations in histone modification in
cancer cells (4). DNA methylation
and histone modifications are interlinked via methyl-CpG-binding
proteins (MeCPs), among which MeCP2 has an important role in
establishing this interaction (5).
MeCPs mediate transcriptional repression in a sequence-independent
process involving the modification of the chromatin structure and
histone acetylation levels (6,7).
Recent studies have shown that MeCP2 is involved in several human
cancers types, including breast cancer (8), hepatocellular carcinoma (9), osteosarcoma (10) and endometrial cancer (11). Darwanto et al (12) revealed that MeCP2 was highly
expressed in well-differentiated adenocarcinoma and mucinous
adenocarcinoma tissues compared with normal and other tumorous
tissues, particularly at the site of invasion of colorectal cancer
tissues. The accumulation of a large number of CpG loci a at the
5′-flanking region of the MeCP2 gene suggests that epigenetic
events may be involved in the regulation of the observed periodic
plasticity of MeCP2 expression during cancer progression. Pancione
et al (13) demonstrated
in vivo as well as in vitro that MeCP2, upon its
recruitment, causes transcriptional silencing of peroxisome
proliferator-activated receptor γ (PPARG) during colon
tumorigenesis via exerting repressive effects on chromatin
signatures, resulting in an increased cell-proliferative and
invasive potential of colorectal cancer. To the best of our
knowledge, the functional role of MeCP2 in the proliferation and
migration of colorectal cancer cells has remained to be
elucidated.
RNA interference (RNAi)-mediated gene silencing is a
potential therapeutic strategy, which has been evaluated in
clinical trials for a number of diseases (14,15).
Due to their minimal toxicity and ability of stable transgene
expression, lentiviral vectors are among the most promising
vehicles for efficient gene delivery in basic research as well as
gene therapy (16,17). The present study assessed the role
of MeCP2 in colorectal cancer and silenced MeCP2 by
lentivirus-mediated RNA interference in colorectal cancer cells to
assess its effects on cell proliferation, the cell cycle and
migration in vitro. The present study suggested that MeCP2
is a potential target for gene therapy in colorectal cancer.
Materials and methods
Cell lines and cell culture
The HCT116, DLD-1, SW480, LoVo, SW1116 and SW620
human colorectal cancer cell lines and the 293T human embryonic
kidney cell line were obtained from the Cell Bank of the Chinese
Academy of Science (Shanghai, China). HCT116, DLD-1, SW480 and LoVo
cells were cultured in RPMI-1640 (Hyclone, Logan, UT, USA)
containing 10% fetal calf serum (Biowest, Nuaillé, France). SW620
cells were cultured in L-15 medium (Sigma-Aldrich, St. Louis, MO,
USA) containing 10% fetal calf serum. SW1116 and 293T cells were
cultured in DMEM (Hyclone) containing 10% fetal calf serum.
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
The expression levels of MeCP2 in the DLD-1, HCT116,
SW1116, SW620, SW480 and LoVo colorectal cancer cell lines were
determined using RT-qPCR (CFX96; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). First, total RNA was extracted using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA)
and subjected to reverse transcription using Moloney murine
leukemia virus reverse transcriptase (Promega Corp., Madison, WI,
USA) according to the manufacturer's instructions. Subsequently 5
μl of the resulting cDNA was amplified by PCR in a final
volume of 20 μl containing 0.8 μl primers (2.5
μM) and 10 μl SYBR premix exTaq (Takara, Dalian,
China). The thermocycling conditions were as follows: 95°C for 1
min followed by 40 cycles of 95°C for 5 sec and 60°C for 20 sec,
with the absorbance value read at the extension stage. Primers with
the following sequences were used for RT-qPCR: MeCP2 forward,
5′-AGC AGT GAG AGC AGA TGA GGT G-3′ and reverse, 5′-GCC CAG GAT AGA
GGA GAC AAA GC-3′; β-actin forward, 5′-GTG GAC ATC CGC AAA GAC-3′
and reverse, 5′-AAA GGG TGT AAC GCA ACT A-3′ (Genewiz, Inc.,
Suzhou, China). Data analysis was performed using the
2−ΔΔCt method (18).
The assay was performed as three independent experiments.
Western blot analysis
To prepare protein extracts, DLD-1, HCT116, SW1116,
SW620, SW480 and LoVo cells were scraped on the ice, collected by
centrifugation (12,000 × g, 15 min, 4°C) and incubated with freshly
prepared 2X SDS lysis buffer [4% SDS (Sangon Biotech Co., Ltd.,
Shanghai, China), 200 mM NaCl (Sangon Biotech Co., Ltd), 10%
glycerol (Sangon Biotech Co., Ltd.), 100 mM Tris (pH 6.8; Sangon
Biotech Co., Ltd.) and 2 mM EDTA (Sangon Biotech Co., Ltd.)] for 10
min. Following centrifugation, the protein concentration in the
supernatant was determined using a BCA Protein Assay kit (Beyotime
Institute of Biotechnology, Haimen, China). Equal amounts of
protein (30 μg per experimental group) were boiled for 10
min in loading buffer [250 mM Tris-HCl (pH 6.8), 10% w/v SDS, 0.5%
w/v bromophenol blue (Sangon Biotech Co., Ltd.), 50% v/v glycerol,
5% w/v β-mercaptoethanol (Sangon Biotech Co., Ltd.)] prior to
separation by 10% SDS-PAGE where samples were linearized at 80 V
for 30 min, then separated at 120 V for 90 min. Subsequently,
electrotransfer to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA) was conducted at 300 mA for 1.5 h.
Membranes were blocked in Tris-buffered saline containing Tween 20
(TBST) with 5% skimmed milk. Subsequently, the membranes were
incubated with rabbit anti-human polyclonal MeCP2 (1:500;
Proteintech Group, Inc., Chicago, IL, USA; cat. no. 10861-1-AP) and
rabbit anti-human polyclonal GAPDH (1:3,000; Proteintech Group,
Inc., Chicago, IL, USA; cat. no. 10494-1-AP) antibodies overnight
at 4°C, followed by incubation with the secondary goat anti-rabbit
IgG antibody (1:5,000; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA; cat. no. sc-2054) for 2 h at room temperature. The membrane
was washed three times with TBST prior to each step. Protein bands
were visualized using the ECL prime™ blotting system (GE
Healthcare, Little Chalfont, UK).
Lentiviral packaging vector
The lentiviral backbone plasmid pFH-L as well as the
pHelper plasmids pVSVG-I and pCMVΔR8.92 were purchased from
Shanghai Hollybio Co. Ltd., (Shanghai, China). pFH-L containing RNA
polymerase III promoter H1 initiates the expression of the inserted
small hairpin (sh)RNA sequence, which continuously silences MeCP2
in the host cells. In addition, pFH-L expresses the reporter gene
green fluorescent protein GFP with activation via the
cytomegalovirus promoter.
Construction of lentiviral vectors
Short-hairpin RNAs (shRNAs) specifically targeting
human MeCP2 were designed based on the GenBank information for
MeCP2 (ID, NM_004992.3). The shRNA targeting human MeCP2 (shMeCP2,
5′-GCC GTG AAG GAG TCT TCT ATC CTC GAG GAT AGA AGA CTC CTT CAC
GGC-3′) and the negative control shRNA (shCon, 5′-GCG GAG GGT TTG
AAA GAA TAT CTC GAG ATA TTC TTT CAA ACC CTC CGC TTT TTT-3′) were
designed and synthesized by Shanghai Hollybio Co. Ltd. (Shanghai,
China). Following annealing of the two oligos in annealing buffer
[10 mM Tris (pH 8.0), 50 mM NaCl and 1 mM EDTA] at 60°C for 20 sec,
the resulting duplex DNAs were cloned into the lentiviral vector
pFH-L (Shanghai Hollybio) and transfected into competent DH5α E.
coli cells (Tiangen Biotech Co., Ltd., Beijing, China). DNA
sequencing was used to verify the positive clones.
For recombinant amplification of the vectors, 293T
cells were co-transfected with MeCP2 RNAi lentiviral expression
vector and control vector with the packaging vectors pVSVG-I and
pCMVΔR8.92 (Shanghai Hollybio) following the manufacturer's
instructions for Lipofectamine 2000 (Invitrogen). Following 48 h of
transfection, supernatants containing the lentiviruses Lv-shCon or
Lv-shMeCP2 were harvested, which were purified using
ultracentrifugation prior to determination of the lentiviral
titer.
Lentiviral transfection
DLD-1 cells in the logarithmic growth phase were
seeded into six-well plates at 5×104 per well and
cultured overnight. The lentiviruses were transfected into the
cells at a multiplicity of infection of 60. Successfully
transfected cells were identified by detection of GFP using a
fluorescent microscope (BX50; Olympus Corporation, Tokyo, Japan),
with the percentage of GFP-positive cells representing the
transfection efficiency. Cells were harvested at day four of
transfection and the knockdown efficiency of MeCP2 was evaluated by
RT-qPCR and western blot analysis.
Proliferation assay
DLD-1 cells were harvested at day four of
transfection and seeded into a 96-well plate at a density of
3×103 cells/well in triplicate. Following incubation for
1, 2, 3, 4 or 5 days with the media replaced every other day, cells
were subjected to the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl
tetrazolium bromide (MTT) assay. In brief, following addition of 20
μl MTT solution (5 mg/ml; Sigma-Aldrich) to each well,
plates were incubated at 37°C for 4 h. In order to dissolve the
generated formazan crystals, wells were incubated with 100
μl acidic isopropanol (10% SDS, 5% isopropanol and 0.01
mol/l HCl) at 37°C for 10 min with agitation. The absorbance of
each well at 595 nm was determined using a microplate reader
(Epoch; BioTek, Winooski, VT, USA). The assay was performed as
three independent experiments.
Colony formation assay
DLD-1 cells were harvested at day four of
transfection. Cells were seeded onto six-well plates at a density
of 500 cells/well and allowed to form colonies over 11 days. Cell
colonies were fixed in 4% paraformaldehyde and stained with crystal
violet (Beyotime Institute of Biotechnology). Colonies (>50
cells) were counted directly on the plate using a microscope. At
least three independent experiments were performed to determine the
number of colonies.
Cell cycle analysis
Following seven days of transfection, DLD-1 cells
were harvested by trypsinization. Following suspension in
phosphate-buffered saline (PBS), the cells were centrifuged (1,000
× g, 5 min, 4°C) and fixed in 70% ethanol at 4°C for 1 h. Following
two washes with PBS, cells were re-suspended in 1 ml PBS containing
500 U/ml RNase (Nanjing KeyGen Biotech. Co., Ltd., Nanjing, China)
and incubated for 30 min at 37°C. Subsequently, cells were
incubated with 20 μg/ml propidium iodide (PI; Nanjing KeyGen
Biotech. Co., Ltd.) for 30 min at room temperature in the dark to
stain cellular DNA. Quantification of DNA was performed using a
FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ,
USA) using CellQuest software, version 6.1 (BD Biosciences). A plot
of the PI fluorescence signal at the FL2A peak vs. the cell count
was used to discriminate G2/M cells from G0/G1 doublets. The
relative populations of cells in G0/G1, S and G2/M phases of the
cell cycle were determined.
Migration assays
DLD-1 cells and Lv-shMeCP2-transfected DLD-1 cells
(1×105 cells/well) in 200 μl FBS-free RPMI 1640
medium were seeded into the upper chambers of Transwell plates
(pore size, 8 μm), while the lower chamber was filled with
800 μl RPMI 1640 containing 10% FBS. After 24 h of
incubation, cells on the upper surface of the membrane were removed
using a cotton swab, and cells which had migrated to the lower side
of the membrane were fixed with 10% methanol, stained with crystal
violet and examined under a microscope. A total of five random
high-power microscopic fields (magnification, 40 ×) per filter were
captured and the number of migrated cells was directly counted.
Subsequently, the crystal violet on the lower membrane was
dissolved in 33% acetic acid and the absorbance of the eluant at
570 nm was determined using the microplate reader (Epoch). Three
independent experiments were performed to determine the number of
migrated cells.
Statistical analysis
SPSS software (version 16.0; SPSS, Inc., Chicago,
IL, USA) was used for statistical analysis. All experiments were
performed in triplicate and values are expressed as the mean ±
standard deviation where applicable. Statistically significant
differences between groups were determined by Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference between values.
Results
MeCP2 expression in colorectal cancer
cell lines
To explore the role of MeCP2 in human colorectal
cancer, the expression levels of MeCP2 in six human colorectal
cancer cells lines, DLD-1, HCT116, SW1116, SW620, SW480 and LoVo,
were determined. As depicted in Fig.
1A, RT-qPCR analysis revealed that MeCP2 mRNA was expressed in
all six colorectal cancer cell lines, with the highest expression
in DLD-1 cells. Furthermore, western blot analysis revealed the
presence of MeCP2 protein in colorectal cancer cells (Fig. 1B). Therefore, DLD-1 cells were
selected for use in the subsequent experiments.
Lv-shMeCP2 efficiently mediates MeCP2
knockdown
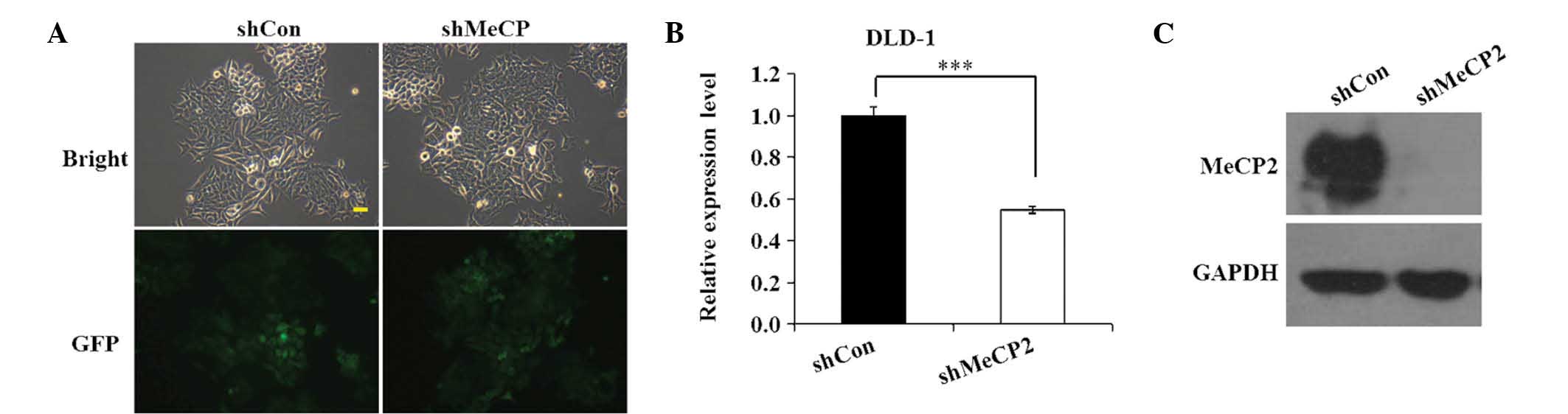
To determine the transfection efficiency of
Lv-shMeCP2 and Lv-shCon in DLD-1 cells, GFP expression was observed
under a fluorescent microscope at 96 h after transfection. As shown
in Fig. 2A, >80% DLD-1 cells
were GFP-positive in the shCon and shMeCP2 groups, indicating that
Lv-shMeCP2- and Lv-shCon were successfully constructed and that
their transfection efficiency was high. Next, RT-qPCR and western
blot analyses were performed to determine the mRNA and protein
levels of MeCP2 in the shCon and shMeCP2 groups. As shown in
Fig. 2B, Lv-shMeCP2 reduced the
expression of MeCP2 mRNA by ~45.2% in DLD-1 cells relative to that
in the shCon group (P<0.001). Western blot analysis validated
that MeCP2 expression in DLD-1 cells relative to GAPDH was knocked
down by transfection with shMeCP2, while that in the shCon group
was still present (Fig. 2C). These
analyses demonstrated that Lv-shMeCP2 efficiently knocked down
MeCP2, which was therefore used in the subsequent experiments.
Knockdown of MeCP2 inhibits colorectal
cancer cell growth
The effects of MeCP2 knockdown on the growth of
colorectal cancer cells in vitro were assessed using MTT and
colony formation assays. The cell proliferation was assessed using
an MTT assay once daily over five days. As shown in Fig. 3A, MeCP2 silencing inhibited DLD-1
cell proliferation in a time-dependent manner. Compared with that
in the shCon group, the cell viability in the shMeCP2 group was
significantly reduced on days four and five (P<0.001).
Furthermore, the colony formation capacity of DLD-1 cells
transfected with Lv-shMeCP2 and Lv-shCon was investigated. As shown
in Fig. 3B, the size of single
colonies was observed using light and fluorescence microscopy. The
sizes as well as the number of single colonies in the shMeCP2 group
were reduced compared with those in the shCon group. As shown in
Fig. 3C, the number of DLD-1 cell
colonies in the shMeCP2 was significantly reduced to 62±5, as
compared with 144±6 in the shCon group (P<0.001). Collectively,
these results demonstrated that knockdown of MeCP2 by
lentivirus-mediated siRNA inhibited the growth of colorectal cancer
cells.
Knockdown of MeCP2 causes cell-cycle
arrest in G0/G1 phase in DLD-1 cells
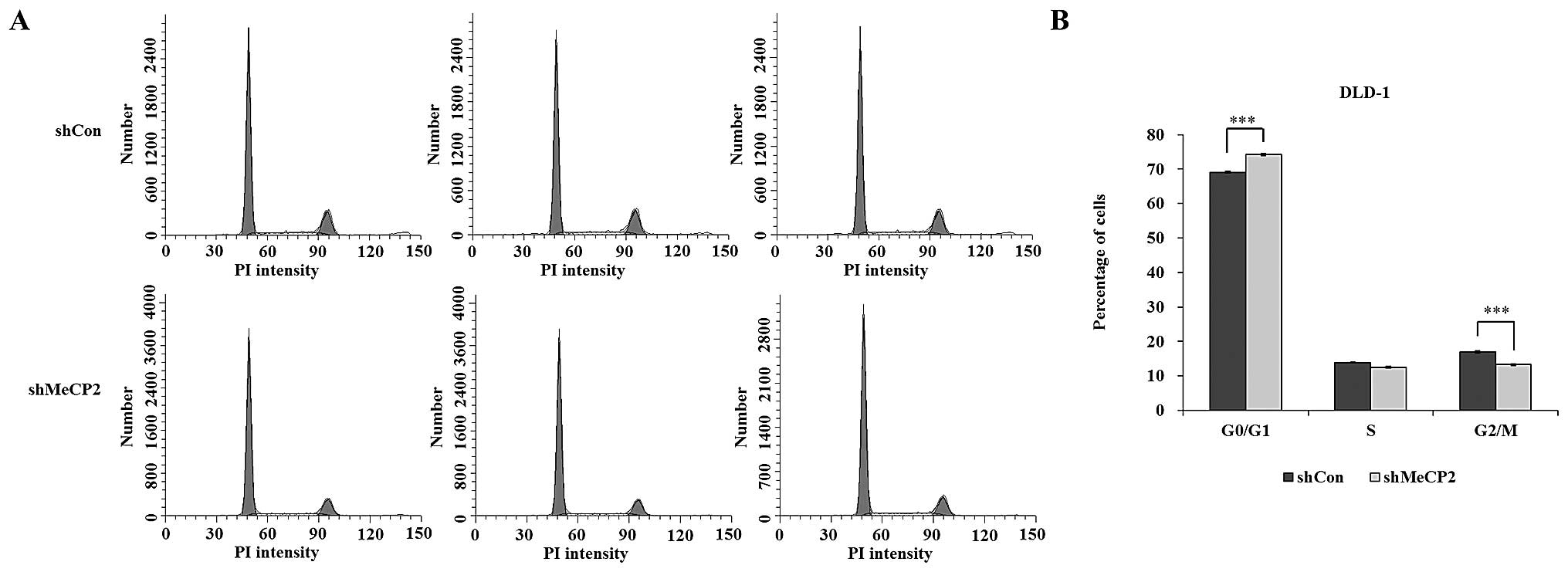
To evaluate the effects of MeCP2 knockdown on the
cell cycle distribution, PI staining followed by flow cytometric
analysis was performed. The proportion of cells in G0/G1 phase was
74.19±0.31 and 69.12±0.18% in the shMeCP2 and shCon groups,
respectively, while the proportion of cells in S phase was
12.54±0.21 and 13.92±0.14%, G2/M-phase was 13.28±0.29 and
16.96±0.31%, respectively. MeCP2 silencing resulted in a
statistically significant increase in the G0/G1-phase population
(P<0.001) as shown in Fig. 4A and
B. These results indicated that downregulation of MeCP2
expression caused cell-cycle arrest in G0/G1 phase.
Knockdown of MeCP2 reduces DLD-1 cell
migration
In order to explore the effects of MeCP2 knockdown
on DLD-1 cell migration, a Transwell migration assay was performed.
As shown in Fig. 5A–C a large
proportion of cells in the shCon group migrated to the lower
surface of the filter, while the number of migrated cells in the
shMeCP2 group was significantly decreased by 47.6% (P<0.001).
These results indicated that downregulation of MeCP2 inhibited the
migratory ability of colorectal cancer cells.
Discussion
MeCP2 has been detected in the majority of
colorectal cancer tissues, particularly at the invasion site of
cancers (12). To explore the
association between MeCP2 and colorectal tumorigenesis, the
expression of MeCP2 in colorectal cancer cells was inhibited
through lentivirus-mediated RNAi. The results of the MTT assay
performed in the present study indicated that downregulation of
MeCP2 expression inhibited cell proliferation. In line with this
finding, the colony formation assay showed that knockdown of MeCP2
inhibited the colony formation ability of colorectal cancer cells.
These findings indicated that MeCP2 promotes cancer cell growth and
suggested that MeCP2 may have an important role in the early stage
of colon tumor development. To explore the potential underlying
mechanism the role of MeCP2 in the growth of colorectal cancer
cells, DLD-1 cells with stable knockdown of MeCP2 were subjected to
flow cytometric cell cycling analysis. It was found that the
specific downregulation of MeCP2 in DLD-1 cells led to G0/G1-phase
arrest. It is known that in G0/G1 phase, cells do not divide,
increase in size and accumulate nutrients (19). In order to proliferate, cells must
enter S phase for DNA synthesis, while blocking of the G1/S-phase
transition inhibits proliferation. MeCP2 acts as a transcriptional
repressor to control gene expression in mammalian cells, which it
exerts through non-specific binding to methylated CpG islands
(20), thereby preventing DNA
binding of transcription factors such as Sp1, which results in
histone deacetylase-mediated alteration of the chromatin structure
(21). Furthermore, it has been
revealed that upon its overexpression, MeCP2 regulates E-cadherin
(E-cad) expression in colorectal cancer; with ongoing tumor
progression, loss of E-cad expression was shown to lead to the
de-differentiation of human carcinomas in vitro and in
vivo (12). Due to the
critical function of MeCP2 on E-cad expression, it can be
hypothesized that the observed reduction of cell proliferation
following MeCP2 knockdown may be due to the inhibition of
de-differentiation through loss of E-cad suppression.
Colorectal cancer is a highly metastatic malignancy.
However, the impact of MeCP2 on colorectal cancer-cell migration
has not been studied in detail. Therefore, the present study
examined the effects of MeCP2 knockdown on the migration of DLD-1
cells using a Transwell migration assay. The results demonstrated
that downregulation of MeCP2 expression in DLD-1 cells markedly
reduced their migration capacity in vitro. These results
corresponded to a recent study reporting that MeCP2 silencing
inhibited osteosarcoma cell proliferation, migration and invasion
(10). The E-cad gene encodes a
cell-surface adhesion protein that has a crucial role in homotypic
cell-cell adhesion and maintenance of epithelial morphology, while
loss of E-cad expression and function inhibits cell-cell adhesion,
thereby inducing tumor-cell invasion and metastasis (12). Therefore, MeCP2 depletion in the
present study was likely to have inhibited the loss of E-cad
expression, which led to the observed reduction of colorectal
cancer-cell migration.
Although the underlying molecular mechanisms of the
effects of MeCP2 gene silencing have not been clearly demonstrated,
the present study evidenced that MeCP2 knockdown was capable of
inhibiting colorectal cancer cell proliferation and migration.
Thus, it is speculated that MeCP2 may be a molecular therapeutic
target in the treatment of colorectal cancer.
In conclusion, the present study demonstrated that
lentivirus-mediated RNAi with MeCP2 expression significantly
inhibited colorectal cancer cell proliferation and migration.
Therefore, MeCP2 represents a novel molecular target for the
treatment of colorectal cancer.
Acknowledgments
The authors are grateful for the financial support
from the Natural Science Foundation of Shanghai (no.
12ZR1404200).
References
|
1
|
Wang WS, Chen PM and Su Y: Colorectal
carcinoma: From tumorigenesis to treatment. Cell Mol Life Sci.
63:663–671. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li Z, Tian T, Hu X, Zhang X, Li L, Nan F,
Chang Y, Wang X, Sun Z, Lv F and Zhang M: Targeting Six1 by
lentivirus-mediated RNA interference inhibits colorectal cancer
cell growth and invasion. Int J Clin Exp Pathol. 7:631–639.
2014.PubMed/NCBI
|
|
3
|
Jones PA and Baylin SB: The epigenomics of
cancer. Cell. 128:683–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esteller M: Epigenetics in cancer. N Engl
J Med. 358:1148–1159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hite KC, Adams VH and Hansen JC: Recent
advances in MeCP2 structure and function. Biochem Cell Biol.
87:219–227. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bestor TH: Gene silencing. Methylation
meets acetylation. Nature. 393:311–312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nan X, Ng HH, Johnson CA, Laherty CD,
Turner BM, Eisenman RN and Bird A: Transcriptional repression by
the methyl-CpG-binding protein MeCP2 involves a histone deacetylase
complex. Nature. 393:386–389. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ray BK, Dhar S, Henry C, Rich A and Ray A:
Epigenetic regulation by Z-DNA silencer function controls
cancer-associated ADAM-12 expression in breast cancer: Cross-talk
between MeCP2 and NF1 transcription factor family. Cancer Res.
73:736–744. 2013. View Article : Google Scholar
|
|
9
|
Zhao LY, Zhang J, Guo B, Yang J, Han J,
Zhao XG, Wang XF, Liu LY, Li ZF, Song TS and Huang C: MECP2
promotes cell proliferation by activating ERK1/2 and inhibiting p38
activity in human hepatocellular carcinoma HEPG2 cells. Cell Mol
Biol (Noisy-le-grand). (Suppl 59): OL1876–OL1881. 2013.
|
|
10
|
Meng G, Lv Y, Dai H, Zhang X and Guo QN:
Epigenetic silencing of methyl-CpG-binding protein 2 gene affects
proliferation, invasion, migration and apoptosis of human
osteosarcoma cells. Tumour Biol. 35:11819–11827. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chu Y, Wang Y, Zhang G, Chen H, Dowdy SC,
Xiong Y, Liu F, Zhang R, Li J and Jiang SW: Chromatin composition
alterations and the critical role of MeCP2 for epigenetic silencing
of progesterone receptor-B gene in endometrial cancers. Cell Mol
Life Sci. 71:3393–3408. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Darwanto A, Kitazawa R, Maeda S and
Kitazawa S: MeCP2 and promoter methylation cooperatively regulate
E-cadherin gene expression in colorectal carcinoma. Cancer Sci.
94:442–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pancione M, Sabatino L, Fucci A, Carafa V,
Nebbioso A, Forte N, Febbraro A, Parente D, Ambrosino C, Normanno
N, et al: Epigenetic silencing of peroxisome proliferator-activated
receptor γ is a biomarker for colorectal cancer progression and
adverse patients' outcome. PLoS One. 5:e142292010. View Article : Google Scholar
|
|
14
|
Lin HC, Wu CL, Chen YL, Huang JS, Wong TY
and Yuan K: High-level β1-integrin expression in a subpopulation of
highly tumorigenic oral cancer cells. Clin Oral Investig.
18:1277–1284. 2014. View Article : Google Scholar
|
|
15
|
Ren W, Wang X, Gao L, Li S, Yan X, Zhang
J, Huang C, Zhang Y and Zhi K: miR-21 modulates chemosensitivity of
tongue squamous cell carcinoma cells to cisplatin by targeting
PDCD4. Mol Cell Biochem. 390:253–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu LL, Chang K, Lu LS, Zhao D, Han J,
Zheng YR, Yan YH, Yi P, Guo JX, Zhou YG, et al: Lentivirus-mediated
RNA interference targeting the H19 gene inhibits cell proliferation
and apoptosis in human choriocarcinoma cell line JAR. BMC Cell
Biol. 14:–26. 2013. View Article : Google Scholar
|
|
17
|
Sun W, Yao L, Jiang B, Guo L and Wang Q:
Spindle and kinetochore-associated protein 1 is overexpressed in
gastric cancer and modulates cell growth. Mol Cell Biochem.
391:167–174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST©) for group-wise
comparison and statistical analysis of relative expression results
in real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar
|
|
19
|
Zhang S, Yang X, Shi H, Li M, Xue Q, Ren
H, Yao L, Chen X, Zhang J and Wang H: Overexpression of leucine
aminopeptidase 3 contributes to malignant development of human
esophageal squamous cell carcinoma. J Mol Histol. 45:283–292. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nan X, Campoy FJ and Bird A: MeCP2 is a
transcriptional repressor with abundant binding sites in genomic
chromatin. Cell. 88:471–481. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kudo S: Methyl-CpG-binding protein MeCP2
represses Sp1-activated transcription of the human leukosialin gene
when the promoter is methylated. Mol Cell Biol. 18:5492–5499. 1998.
View Article : Google Scholar : PubMed/NCBI
|