Introduction
Renal cell carcinoma (RCC) is the third most common
type of urological cancer, and accounts for 2% of adult
malignancies (1). RCC is composed
of several subtypes, including clear cell, chromophobe, papillary
and collecting duct carcinoma (2),
categorized based on their clinical outcome and biological
features. Clear cell RCC is the most common subtype, and accounts
for ~80% of cases of RCC (3). For
all stages combined, the five-year relative survival rate is 55%
(4). Despite the rapid advancement
in the diagnostic and therapeutic strategies for RCC, the mortality
rate has not changed significantly (5). Surgical resection remains the only
definitive treatment for RCC, however, 20–40% patients develop
recurrence following curative nephrectomy (6). Therefore, it is crucial to
investigate the molecular mechanism underlying the progression of
RCC. MicroRNAs (miRNAs/miRs) may offer potential applications for
the diagnosis, prognosis and treatment of RCC.
miRNAs are identified as an abundant class of small,
non-coding RNAs, which are important in the post-transcriptional
regulation of various biological processes (7–9).
Currently, >1,000 miRNAs have been identified in humans.
Generally, miRNAs bind to the 3′ untranslated region (3′-UTR) of
their target genes through imperfect complementation, and repress
gene expression either by increasing mRNA degradation or by
inhibiting translation (10).
miRNAs suppress protein translation by binding to complementary
sequences, which are predominantly located in the 3′-UTR of target
messenger RNA (mRNA) (11,12). miRNAs regulate the expression
levels of genes involved in several cellular processes, including
cell cycle (13,14), differentiation (15,16)
and apoptosis (17,18). Furthermore, each miRNA can
potentially regulate hundreds of mRNAs, and over one third of human
genes may be miRNA targets (19–21).
There is considerable evidence indicating that
miRNAs are involved in human cancer (22–24).
Furthermore, it has also been suggested that deregulated miRNAs may
have a tumor-suppressive or oncogenic role in different types of
cancer by repressing the expression of oncogenes or tumor
suppressor genes (25). Therefore,
the functional identification of miRNAs has become an important
area of investigation in biomedical science, however, the majority
of these miRNAs have unknown functions (26). Massively parallel sequencing
studies have revealed the aberrant expression of miR-362-3p in RCC
(27,28). Our previous study demonstrated that
the expression levels of miR-362-3p are significantly downregulated
in RCC (29). However, the
mechanism and the association between the expression of miR-362-3p
and RCC remain to be fully elucidated. The biological role of
miR-362-3p in RCC also requires further clarification.
The aim of the present study was to analyze the
expression pattern of miR-362-3p in clinical RCC tissue samples,
and examine the effects of miR-362-3p on the proliferation,
migration, invasion, cell cycle and apoptosis of RCC cell lines. In
order to better understand the regulatory mechanism of miR-362-3p,
the nemo-like kinase (NLK) target gene was validated using a
luciferase reporter assay and western blot analysis.
Materials and methods
RCC clinical specimens
A total of 47 patients with RCC underwent routine
surgery at the Zhujiang Hospital, Southern Medical University
(Guangzhou, China) between May 2013 and April 2014. RCC tissue
samples and corresponding adjacent normal renal tissue samples were
collected from these 47 patients, and were immediately snap frozen
in liquid nitrogen, and stored at −80°C until RNA extraction. The
47 patients recruited in the present study included 34 men and 13
women, with a median age of 50 years (range, 24–84 years). None of
the patients had undergone chemotherapy or radiotherapy prior to
surgery. The clinical diagnosis and histology of the tumors from
the patients with RCC was performed by the Cancer Center of the
Southern Medical University. All tissue specimens were acquired on
the basis of their availability for research objective and
following a protocol approved by the ethics committee of Shenzhen
Second People's Hospital (Shenzhen, China). Written informed
consent was obtained from the patients prior to commencement of the
present study. The clinicopathological information of the patients
is shown in Table I. The disease
stage of each patient was classified or reclassified according to
the American Joint Committee on Cancer staging system (30).
 | Table IClinicopathological characteristics
of the 47 patients with renal cancer recruited in the present
study. |
Table I
Clinicopathological characteristics
of the 47 patients with renal cancer recruited in the present
study.
| Factor | Number of
cases |
|---|
| Gender | |
| Male | 34 |
| Female | 13 |
| Age | |
| ≤50 years | 20 |
| >50 years | 27 |
| Pathology | |
| Clear cell renal
cell carcinoma | 36 |
| Papillary renal
cell carcinoma | 5 |
| Chromophobe renal
cell carcinoma | 6 |
| Edmondson
garaging | |
| I+II | 30 |
| III+IV | 17 |
| Tumor size | |
| <7 cm | 39 |
| ≥7 cm | 8 |
| Lymphatic
invasion | |
| Positive | 0 |
| Negative | 47 |
| Distant
metastasis | |
| Positive | 0 |
| Negative | 47 |
| Stage | |
| T1a | 17 |
| T1b | 21 |
| T2 | 6 |
| T3 | 2 |
| T4 | 1 |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
To analyze the expression levels of miR-362-3p,
RT-qPCR was performed using SYBR® Premix Ex Taq™ II
(Perfect Real Time; Takara Bio, Inc., Otsu, Japan) in an Mx3000P
system (Agilent Technologies, Inc., Santa Clara, CA, USA). Tissue
samples were homogenized and RNA was isolated using RNAios Plus
total RNA extraction reagent (Takara Bio, Inc.), according to the
manufacturer's protocol. Reverse transcription into cDNA was
performed using a Primescript™ RT Reagent kit using gDNA Eraser
(Perfect Real Time; Takara Bio, Inc.). U6 was used to normalize the
data. Total RNA was reverse transcribed using the corresponding RT
primer and the TaqMan MicroRNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc. Waltham, MA, USA). The
PCR primer for mature miR-362-3p was designed using a publicly
available databases (miRBase; http://www.mirbase.org/) synthesized by Thermo Fisher
Scientific, Inc. with the following sequences: miR 362-3p forward,
5′-AAC ACA CCT ATT CAA GGA TTC A-3′ and reverse, 5′-ACG TGA CAC GTT
CGG AGA ATT-3′. The expression levels (2−ΔΔCq) were
normalized to those of U6 (U6 sense strand, 5′-CTC GCT TCG GCA GCA
CA-3′, and anti-sense strand, 5′-ACG CTT CAC GAA TTT GCG T-3′). The
2−ΔΔCQ method was used to analyze the data, with the
data presented as log2 (cancer/normal) (31).
Cell culture and miRNA transfection
ACHN and 786-O human renal carcinoma cell lines were
purchased from the American Type Culture Collection (Manassas, VA,
USA). Pairs of synthetic chemically modified short single or
double-stranded RNA oligonucleotides, miR-362-3p mimics and an
appropriate negative control (NC) were purchased from GenePharma
Co. Ltd. (Shanghai, China) with the following sequences: miR 362-3p
mimic strand, 5′-AAC ACA CCT ATT CAA GGA TTC A-3′ and miR 362-3p
negative control sense strand, 5′-TTC TCC GAA CGT GTC ACG TTT-3′,
and anti-sense strand, 5′-ACG TGA CAC GTT CGG AGA ATT-3′.
Transfection was performed using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. The stable ACHN human renal cancer
cell lines and 786O cell lines were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% fetal bovine serum (FBS), 50 µg/ml
streptomycin, 50 U/ml penicillin and 2 mmol/l glutamine
(Invitrogen). The cells were maintained in a 5%
CO2-humidified atmosphere at 37°C. The cells
(~8×103 cells/well) were seeded into a 96-well plate 24
h prior to transfec tion, following which the ACHN and 786-O cells
were transfected with the indicated quantities of the miR-362-3p
mimics or miR-NC using Lipofectamine® reagent, according
to the manufacturer's protocol.
Cell proliferation assay
Cell proliferation was detected using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (5 mg/ml; Sigma-Aldrich, St. Louis, MO, USA). The
miR-362-3p-transfected cells and miR-NC-transfected renal cancer
cells were harvested and dissociated into single-cell suspensions
by digestion with 0.9% trypsin for 3 min at 37°C, followed by
pipetting for 30 times. Cell proliferation was examined at various
time points following transfection (0, 24, 48 and 72 h), with 20
µl MTT (5 mg/ml) added to each well, followed by incubation
for 4 h at 37°C. The MTT medium mixtures were subsequently
discarded, following which 150 µl dimethyl sulf-oxide was
added to each well and agitated for 15 min at room temperature to
solubilize the crystals. The absorbance was measured at a
wavelength of 490 nm (with 630 nm as the reference wavelength)
using an ELISA microplate reader (HR801; Shenzhen Highcreation
Technology Co., Ltd Guangdong, China). All proliferation assays
were repeated as independent experiments at least three times.
Cell migration assay
Cell migration was examined using a wound-healing
assay, as previously described (25). Briefly, the ACHN and 786-O stable
cell lines were plated in 6-well plates (5×105
cells/well) and were grown to 75–90% confluence. The cell
monolayers were then transfected with 50 nm miR-362-3p mimics. A
total of 6 h following transfection, an artificial homogenous wound
was created onto the monolayer using a sterile 200 µl tip.
The cells were subsequently washed with phosphate-buffered saline
(PBS) and maintained in serum-free medium. Images of the cells
migrating into the wound were captured at 0 and 24 h using a
digital camera system (DMIL LED; Leica Microsystems GmbH, Wetzlar,
Germany). MIAS-2000 software (Leica Microsystems GmbH) was used to
determine the migration distance. The experiments were performed
independently in triplicate.
Cell invasion assay
Transwell chambers (8 µm pore size; Corning
Incorporated, Corning, NY, USA) were coated with Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA) (15 µg/filter). The
cells were seeded (5×105 cells/well) in serum-free
medium in the upper chamber (Biocoat Matrigel invasion chamber) and
the lower wells were filled with 10% FBS. The cells were allowed to
migrate through the Matrigel to the lower chamber for 24 h.
Following incubation, the cells were removed from the upper surface
of the filter by scraping with a cotton swab. The cells on the
lower surface of the filter were permeabilized using 1% Triton
X-100 (Sigma-Aldrich), stained with crystal violet (Sigma-Aldrich)
and visualized under a microscope DMI 6000B; Leica Microsystems
GmbH). The numbers of cells, which penetrated through the membrane
were determined by counting the mean number of cells in five
randomly-selected high-power fields. The experiments were repeated
as independent experiments at least three times.
Cell apoptosis assay
An Annexin V-fluorescein isothiocyanate (FITC)
Apoptosis Detection kit (Invitrogen) and propidium iodide (PI;
Invitrogen) were used to assess the apoptotic effect of miR-362-3p.
The renal cells of the different treatment groups were suspended at
a concentration of 1×106 cells/ml. The cell suspension
was transferred to a centrifuge tube, centrifuged at 7.9 × g and
washed with PBS. The cells were then resuspended in 500 µl
cold 1X binding buffer (Invitrogen) with 5 µl Annexin
V-FITC, and incubated for 15 min at room temperature in the dark.
The cells were then centrifuged for 5 min to remove the
supernatant. The cells were then resuspended in 500 µl cold
binding buffer with 3 µl PI, incubated for 15 min and
analyzed by flow cytometry (Navi 105; Beckman Coulter, Brea, CA,
USA). Experiments were performed in three independent repeats.
Cell cycle analysis
To examine the effects of the downregulation of
miR-362-3p on the cell cycle, flow cytometry was performed. For
cell cycle analysis, the miR-362-3p-transfected cells were
harvested 48 h post-transfection, and were trypsinized (Invitrogen)
and fixed with ice-cold 95% ethanol (Shanghai ExCell Biology,
Shanghai, China) overnight at 4°C. The fixed cells were stained
with 50 mg/ml PI, treated with 50 mg/ml RNase and then analyzed
using a flow cytometer (BD Pharmingen). Each assay was
independently repeated three times.
Vector construction and luciferase
reporter assay
TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do) and PicTar
(http://pictar.mdc-berlin.de/) were used
to computationally predict the targets of miR-362-3p. To create a
luciferase reporter construct, the 3′-UTR fragment of NLK,
containing putative binding sites for miR-362-3p was inserted
downstream of firefly luciferase, between the
XhoI-NotI restriction sites in the 3′-UTR of the
hRluc gene in the psiCHECKTM-2 luciferase vector (Promega Corp.,
Madison, WI, USA). Mutant 3′-UTR, which carried the mutated
sequence in the complementary site for miR-362-3p, was generated
using the fusion PCR method, and inserted downstream of firefly
luciferase between the XhoI-NotI restriction sites in
the 3′-UTR of the hRluc gene in the psiCHECKTM-2 luciferase vector.
The following sequences were used: NLK 3′-UTR wild-type sequence,
5′-ATA TCA TTC TAA CGG GTG TGT T 3′ and NLK 3′-UTR mutant sequence,
5′-ATA TCA TTC TAA CGC CAC ACA A-3′, and synthesis was performed at
GenePharma Co., Ltd. Cells grown in a 48-well plate were
co-transfected with miR-362-3p and the luciferase reporter
comprising the wild-type or mutant 3′-UTR of the target gene. The
luciferase assay was performed as described previously (32). The cells (5×105 per
group) were co-transfected with miRNAs and 3′-UTR or a mutant
3′-UTR luciferase reporter, in which the potential binding sites
were manually mutated by exchanging the G and T, A and C as a
control vector. At 48 h post-transfection, luciferase activity was
measured using a Dual-Luciferase Assay kit (Promega Corp.) with a
β-counter luminometer (Promega Corp.). Relative luciferase activity
was calculated as the ratio of raw firefly luciferase activity and
renilla luciferase activity.
RNA isolation and RT-qPCR
Total RNA was isolated from the
miR-362-3p-transfected cells using RNAiso Plus total RNA extraction
reagent Takara, Bio, Inc.) according to the manufacturer's
instructions. The following primers were used: NLK, sense
5′-AGCCGTCATTACAGCAAT-3′ and antisense
5′-TATTCTTCGTCCTAGTAGTTCTG-3′; GAPDH, sense
5′-TGGCCTTCCGTGTTCCTAC-3′ and antisense GAPDH
5′-GAGTTGCTGTTGAAGTCGCA-3′. For the detection of NLP or GAPDH mRNA,
a total of 1 µg mRNA was reverse-transcribed into cDNA using
a One Step PrimeScript™ miRNA cDNA Synthesis kit (Takara, Bio,
Inc.). qPCR was performed using SYBR® Premix Ex Taq™ II
(Perfect Real Time; Takara Bio, Inc.) in a Quant Studio™ system
(Agilent Technologies, Inc., Santa Clara, CA, USA), according to
the manufacturer's instructions with U6 or GAPDH as an internal
reference. The 20-µl reaction mixture contained 10 µl
2X QuantiTect SYBR Green PCR Master mix, 2 µl 10X miScript
Universal Primer, 0.4 µl specific microRNA primer, 1
µl cDNA template and RNase-free water. The thermocycling
conditions were as follows: 95°C for 15 min, followed by 40 cycles
of 94°C for 15 sec, 55°C for 30 sec and 72°C for 30 sec. The
expression levels of miR-362-3P and NLK were quantified using the
2−ΔΔCq method. The RT-qPCR primers were synthesized by
Thermo Fisher Scientific, Inc. with the sequences as follows: NLK
forward, 5′-AGC CG TCA TTA CAG CAA T-3′ and reverse, 5′-TAT TCT TCG
TCC TAG TAG TTCTG-3′; GAPDH forward, 5′-TGG CCT TCC GTG TTC CTAC-3′
and reverse, 5′-GAG TTG CTG TTG AAG TCGCA-3′; miR 362-3p forward,
5′-AAC ACA CCT ATT CAA GGA TTC A-3′ and reverse, 5′-ACG TGA CAC GTT
CGG AGA ATT-3′; U6 forward, 5′-CTC GCT TCG GCA GCA CA-3′ and
reverse, 5′-ACG CTT CAC GAA TTT GCG T-3′.
Western blot analysis
The miR-362-3p-transfected cultured cells were
harvested, and total protein was extracted using
radioimmunoprecipitation assay lysis buffer supplemented with
proteinase/phosphatase inhibitors (Thermo Fisher Scientific, Inc.).
The protein concentration was determined and equilibrated using a
Pierce Bicinchoninic Acid Protein Assay kit (Thermo Fisher
Scientific, Inc.). Equivalent quantities of protein (30 µg)
were separated by 10% SDS-PAGE (Invitrogen) and transfected onto
pre-wetted polyvinylidene fluoride membranes (Millipore, Billerica,
MA, USA). The membrane was blocked in 5% non-fat milk for 1 h and
incubated overnight at 4°C with primary antibody. On the following
day, the membranes were washed three times in Tris-buffered saline
with Tween 20 (TBS-T; Beijing Solarbio Science Technology Co.,
Ltd., Beijing, China) and incubated with horseradish
peroxidase-conjugated secondary antibody in TBS-T for 1 h at room
temperature. An enhanced chemiluminescence kit (Millipore) was used
to detect the secondary antibody. The expression levels of NLK were
analyzed using mouse polyclonal anti-NLK antibody (cat. no.
sc-48361; dilution 1:1,000; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA), and rabbit anti-mouse polyclonal anti-β-tubulin antibody
(cat. no. ab59680; dilution 1:10,000; Abcam, Cambridge, MA, USA)
was used to detect the expression levels of β-tubulin, which was
used as endogenous control to normalize the expression levels of
NLK.
Statistical analysis
The data are presented as the mean ± standard
deviation. Data analysis was performed using Student's t-test to
determine the significance between two variables. The results were
analyzed using SPSS 12.0 (SPSS, Inc., Chicago, IL, USA). P<0.05
was considered to indicate a statistically significant difference.
All experiments were performed in triplicate.
Results
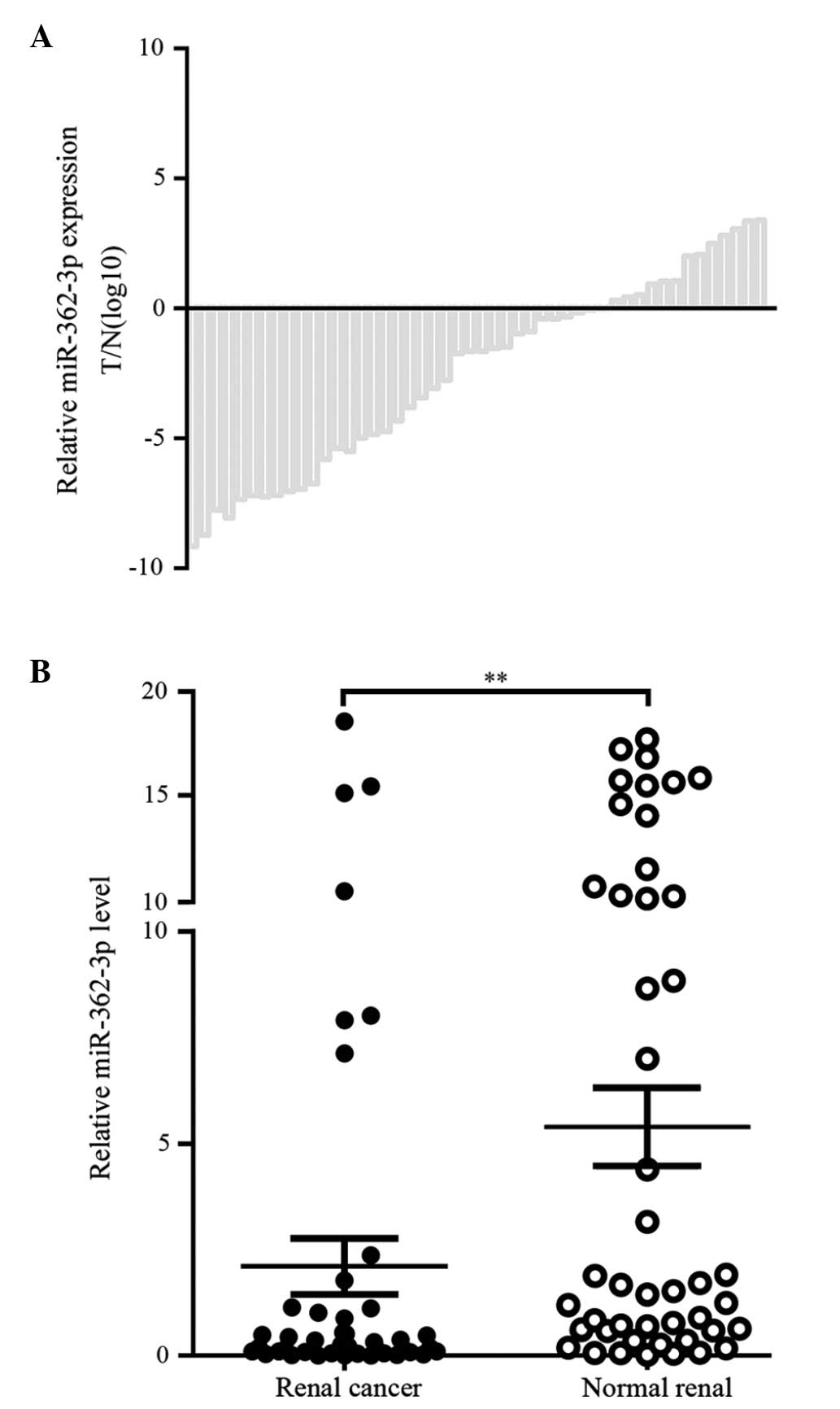
Expression of miR-362-3p is downregulated
in RCC
To investigate the role of miR-362-3p in RCC
development, the expression levels of miR-362-3p were examined in
47 pairs of RCC and adjacent normal kidney tissue samples using
RT-qPCR. The decrease in the expression of miR-362-3p in the RCC
tissue samples was more marked, compared with that in the adjacent
normal kidney tissue samples (Fig. 1A
and B). The expression levels of miR-362-3p were decreased in
35 tissue samples (74.5%), with an overall average of 44.9%
downregulation (P= 0.005). These results suggested that miR-362-3p
may act as tumor suppressor in RCC.
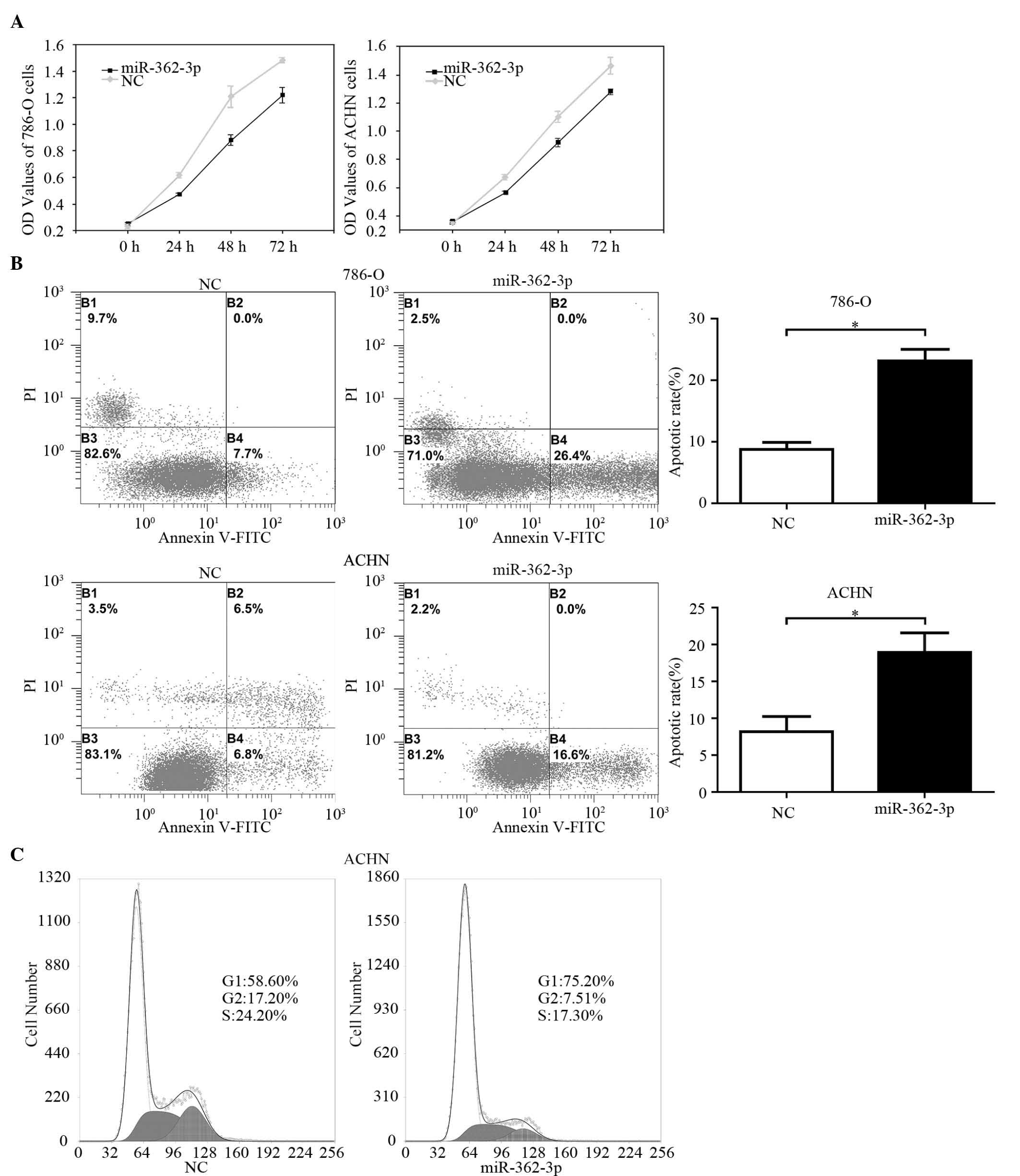
Effects of miR-362-3p on cell
proliferation, apoptosis and cell cycle in RCC cell lines
To assess the biological role of miR-362-3p in RCC,
miR-362-3p mimics or NC were transfected into 786-O and ACHN cells.
The MTT assay demonstrated that the relative cell proliferation in
the miR-362-3p-transfected cells decreased significantly by 23.1%
(24 h), 27.1% (48 h) and 17.6% (72 h) in the 786-O cells
(P<0.001). In the ACHN cells, the inhibition rates of cellular
proliferation were 23.2% (24 h), 20.2% (48 h) and 14.6% (72 h),
respectively (P<0.001; Fig.
2A).
The rates of apoptosis in the 786-O and ACHN cells
were analyzed using flow cytometry using miR-362-3p mimics or NC
(Fig. 2B). As shown in Fig. 2B, miR-362-3p significantly promoted
apoptosis of the 786-O cells, compared with the NC (24.9, vs. 7.9%,
respectively; P=0.003) and of the ACHN cells (18.3, vs. 7.85%,
respectively; P=0.021).
Inhibition of cell growth in cancer cells is
generally associated with cell cycle arrest. Therefore, a cell
cycle assay was performed using flow cytometry with the miR-362-3p
mimics or NC (Fig. 2C). A
significant increase in the percentage of cells in the
G1 phase to 75.2%, was observed in the
miR-362-3p-transfected renal cancer cells, compared with 58.6% in
the miR-NC-transfected cells. These results suggested that
miR-362-3p may lead to G1 cell cycle arrest in RCC
cells.
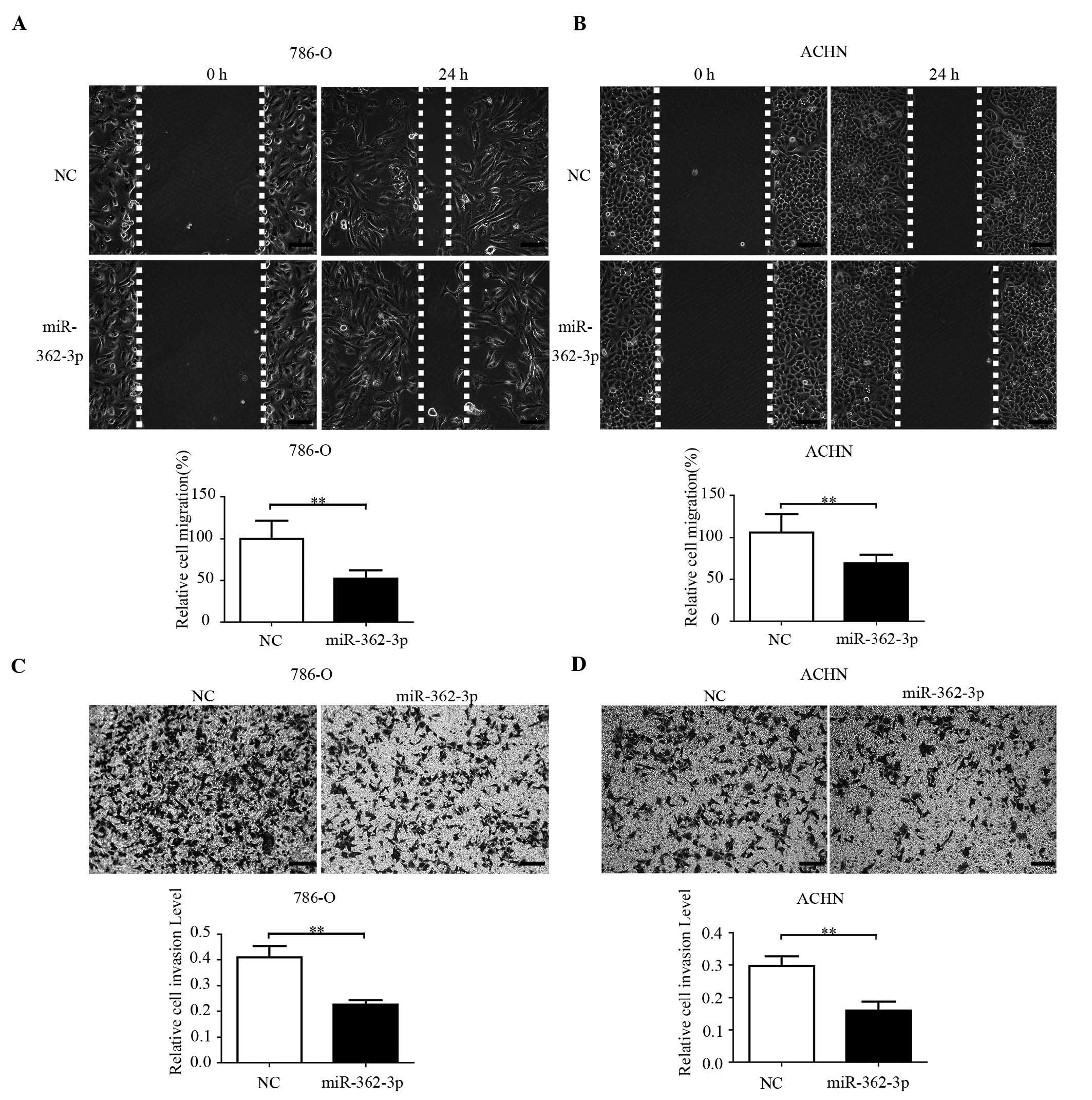
Effects of miR-362-3p on cell migration
and invasion in RCC cell lines
To investigate whether miR-362-3p had an effect on
facilitating renal cancer cell migration, migration was assessed
using a wound healing assay in the 786-O and ACHN cells. As shown
in Fig. 3A and B, cell migration
was significantly reduced in the cells transfected with miR-362-3p,
compared with those in the miR-NC group (40 and 50%, respectively;
P<0.05; Fig. 3A and B). These
results suggested that miR-362-3p has a negative effect on cellular
migration.
As invasion is an important characteristic of
malignant tumors, the present study investigated the effects of
miR-362-3p on tumor invasion in the 786-O and ACHN cell lines. The
invasion assay demonstrated that cell invasion was significantly
inhibited in the miR-362-3p-transfected cells, compared with the
NC-transfected cells. The number of invading cells decreased by
49.8% (P<0.001) in the 786-O cells and by 55.3% (P<0.001) in
the ACHN cells, suggesting that miR-362-3p inhibited the invasive
potential of the renal cancer cells (Fig. 3C and D).
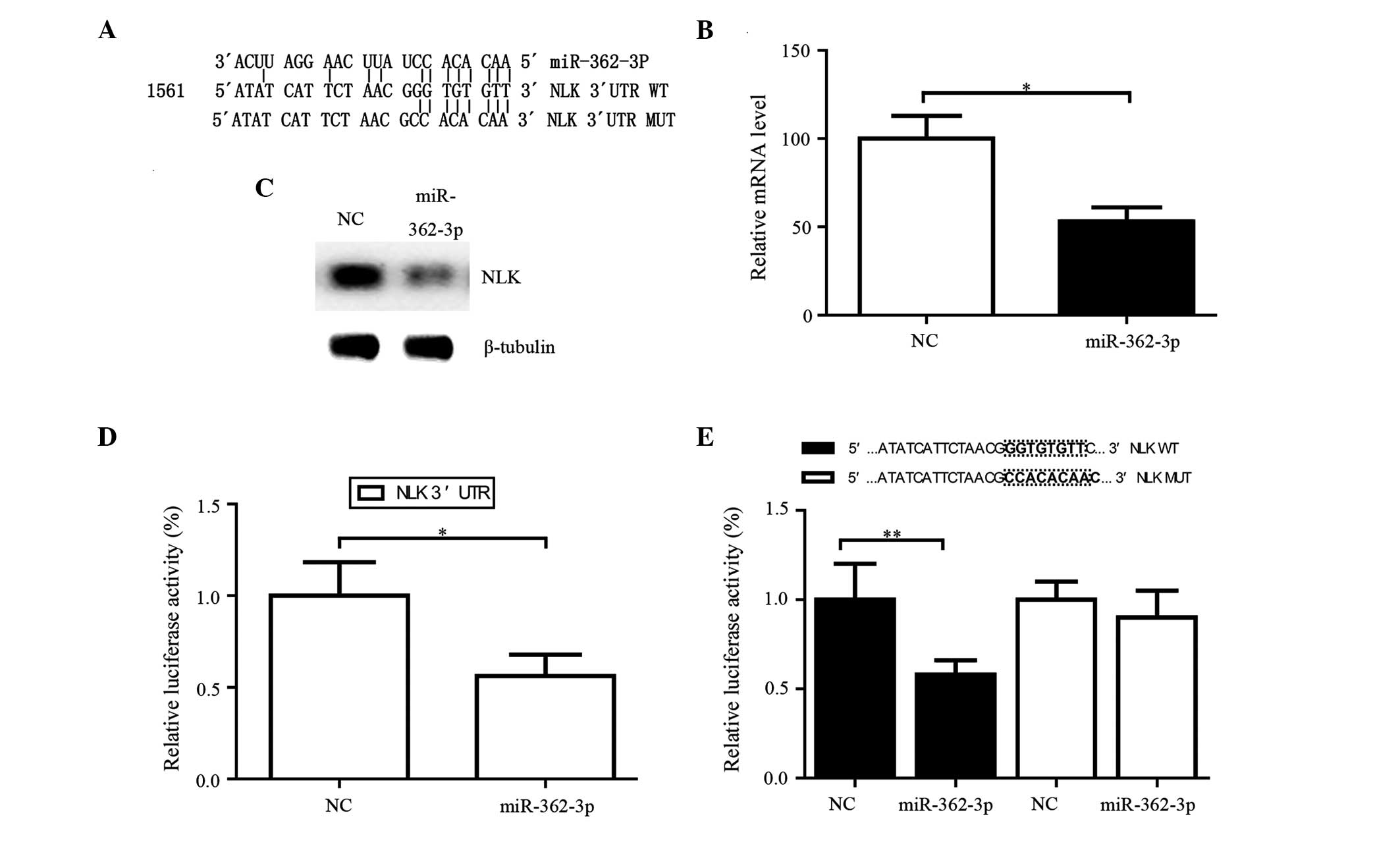
NLK is a target gene of miR-362-3p
TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/microrna/home.do) and PicTar
(http://pictar.mdc-berlin.de/) were used
to computationally predict the targets of miR-362-3p. The
cancer-promoting gene, NLK, was predicted to be one potential
target (Fig. 4A). The potential
binding site of miR-362-3p (position 854–860) was predicted to be
the NLK 3′-UTR. The mRNA and protein expression levels of NLK were
quantified in the renal cancer cells following transfection with
miR-362-3p mimics. The mRNA and protein expression levels of NLK
were decreased following transfection (Fig. 4B and C). As shown in Fig. 4D and E, transfection of the renal
cancer cells with the miR-362-3p mimics in the wild-type 3′-UTR
(pLuc-NLK 3′-UTR-wild) vector significantly reduced luciferase
activity, compared with the NC (P<0.05). However, transfection
of the renal cancer cells with the miR-362-3p mimics in the mutant
3′-UTR (pLuc-NLK 3′-UTR-mut) vector had no effect on luciferase
activity, compared with the control inhibitor (P>0.05). These
results indicated that miR-362-3p downregulated the luciferase
activity of the reporter. The data also identified NLK as a target
gene of miR-362-3p, and identified the sites of interaction in the
3′-UTR of NLK.
Discussion
Despite significant improvements in cancer therapy,
RCC is relatively resistant to traditional cancer treatments,
including radiotherapy, immunotherapy and chemotherapy (33). Therefore, the identification of
novel, more accurate and prophetic prognostic markers, which are
not currently included in conventional staging systems, aim to
improve the prognosis of patients with RCC. miRNAs are aberrantly
expressed in several types of cancer and appear to have diagnostic
and prognostic significance. Dysregulation of miRNAs is associated
with various human diseases, particularly cancer (21). It has been established that miRNAs
regulate various important cellular processes, including
proliferation, cell cycle, differentiation and tumor formation
(34).
Previous studies have demonstrated the presence of
aberrant miRNA expression in human cancer (25), including RCC (35), a number of which function as tumor
suppressor genes or oncogenes (36). Furthermore, next-generation
sequencing techniques have provided further insights into the
genetic basis of RCC (37).
Consequently, the identification of novel biomarkers, particularly
for patients with RCC is urgently required. In our previous study,
massively parallel sequencing revealed the downregulation of
miR-362-3p in ccRCC (29). The
downregulation of miR-362-3p and its functional analysis in colon
cancer cell lines has also suggested a tumor-suppressing role for
miR-362-3p (38). However, the
association between the expression of miR-362-3p and RCC remains to
be elucidated and requires further clarification. The present study
investigated the downregulation of miR-362-3p in RCC tissues,
compared with normal tissues. Based on the results, it was
hypothesized that miR-362-3p may have an important role as a tumor
sup pressor in kidney cancer. The results demonstrated that the
ectopic expression of miR-362-3p caused inhibition of
proliferation, invasiveness and migration, and increased cell
apoptosis in the RCC cells. These results confirmed the anticancer
effect of miR-362-3p, and provide sufficient evidence to support
the tumor suppressor role of miR- 362-3p in RCC (39).
Although the results of the present study
demonstrated the importance of miR-362-3p as a tumor suppressor in
RCC, the precise molecular mechanisms underlying its function
remain to be elucidated. Mature miRNAs regulate the expression of
target genes at the post-transcriptional level via the degradation
of transcripts and inhibition of translation though binding to the
3′-UTR of target mRNA. To acquire a better understanding of the
tumor suppressive effects of miR-362-3p in renal tumor formation,
the analysis of miR-362-3p-predicted targets was performed using
the following algorithms: MiRanda (http://www.microrna.org/microrna/home.do), PicTar
(http://pictar.mdc-berlin.de/) and
TargetScan (http://targetscan.org/). The binding
sites of miR-362-3p were situated on the NLK 3′-UTR, determined
using a luciferase reporter assay. Furthermore, the results of the
present study demonstrated that overexpression of miR-362-3p
downregulated the mRNA and protein expression levels of NLK.
NLK is a promutogenic nemo-like kinase, and is a
classic mediator of the Wnt/β-catenin signaling pathway (40). NLK belongs to the threonine protein
kinase super family and is important in the apoptosis of cancer
cells. Previous studies have also shown that NLK is critical in
tumor occurrence and development, and is therefore becoming the
subject of current studies. For example, studies have revealed that
the expression levels of NLK in cancer tissue samples are
significantly higher, compared with corresponding normal tissue
samples in hepatocellular carcinoma and ovarian cancer (41,42).
In 2003, Yasuda et al demonstrated that over-expression of
NLK may have targets other than TCF in the induction of apoptosis
in human colon carcinoma cells (43). In 2009, Emami et al
(44) revealed that overexpression
of NLK results in more pronounced induction of apoptosis in
androgen receptor (AR)-expressing LNCaP cells, compared with
AR-negative PC-3 cells. In 2010, Jung et al (45) suggested that the expression level
of NLK in cancer tissue samples are significantly higher, compared
with those in corresponding normal liver tissue samples in
hepatocellular carcinoma, and also demonstrated that NLK may be
involved in promoting the growth of hepatoma cells via promoting
cell cycle progression. In 2011, Cui et al (46) reported that NLK induces apoptosis
in glioma cells via the activation of caspases, therefore, NLK may
serve as a useful independent prognostic indicator for glioma. In
2012, Stevens et al (42)
demonstrated that the expression levels of NLK are higher in normal
ovarian tissue samples, compared with ovarian cancer tissue
samples. In the present study, the data showed that miR-362-3p
regulated the expression of NLK by directly binding to its 3′-UTR,
suggesting that miR-362-3p may have tumor suppressive functions by
regulating oncogenic genes in RCC. The overexpression of miR-362-3p
in RCC may lead to the downregulation of NLK, which subsequently
provides a growth and expansion advantage during renal
carcinogenesis.
In conclusion, the present study demonstrated that
miR-362-3p was frequently reduced in clinical tissue specimens of
RCC. miR-362-3p exerted suppressive effects on tumor proliferation,
migration and invasion by directly or indirectly regulating
targeted genes. To the best of our knowledge, the present study
provided the first evidence that miR-362-3P may act as a tumor
suppressor in RCC. These findings may be pivotal in RCC
oncogenesis, and provide evidence supporting the use of miRNAs as a
novel approach for the detection, prevention and treatment of
RCC.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81170613 and
81270740) and the Shenzhen Basic Research knowledge Innovation
Program (grant no. JCYJ20140416180 323426).
References
|
1
|
Grange C, Collino F, Tapparo M and Camussi
G: Oncogenic micro-RNAs and renal cell carcinoma. Front Oncol.
4:492014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopez-Beltran A, Carrasco JC, Cheng L,
Scarpelli M, Kirkali Z and Montironi R: 2009 update on the
classification of renal epithelial tumors in adults. Int J Urol.
16:432–443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Spronsen DJ, de Weijer KJ, Mulders PF
and De Mulder PH: Novel treatment strategies in clear-cell
metastatic renal cell carcinoma. Anticancer Drugs. 16:709–717.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reeves DJ and Liu CY: Treatment of
metastatic renal cell carcinoma. Cancer Chemother Pharmacol.
64:11–25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Janzen NK, Kim HL, Figlin RA and
Belldegrun AS: Surveillance after radical or partial nephrectomy
for localized renal cell carcinoma and management of recurrent
disease. Urol Clin North Am. 30:843–852. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shao Y, Zhang SQ, Quan F, Zhang PF and Wu
SL: MicroRNA-145 inhibits the proliferation, migration and invasion
of the human TCA8113 oral cancer line. Oncol Lett. 6:1636–1640.
2013.PubMed/NCBI
|
|
8
|
Yang L, Wang YL, Liu S, Zhang PP, Chen Z,
Liu M and Tang H: miR-181b promotes cell proliferation and reduces
apoptosis by repressing the expression of adenylyl cyclase 9 (AC9)
in cervical cancer cells. FEBS Lett. 588:124–130. 2014. View Article : Google Scholar
|
|
9
|
Billeter AT, Barnett RE, Druen D, Polk HC
Jr and van Berkel VH: MicroRNA as a new factor in lung and
esophageal cancer. Semin Thorac Cardiovasc Surg. 24:155–165. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eulalio A, Huntzinger E and Izaurralde E:
Getting to the root of miRNA-mediated gene silencing. Cell.
132:9–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lytle JR, Yario TA and Steitz JA: Target
mRNAs are repressed as efficiently by microRNA-binding sites in the
5′UTR as in the 3′UTR. Proc Natl Acad Sci USA. 104:9667–9672. 2007.
View Article : Google Scholar
|
|
12
|
Wightman B, Bürglin TR, Gatto J, Arasu P
and Ruvkun G: Negative regulatory sequences in the lin-14
3′-untranslated region are necessary to generate a temporal switch
during Caenorhabditis elegans development. Genes Dev. 5:1813–1824.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lal A, Navarro F, Maher CA, Maliszewski
LE, Yan N, O'Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O,
et al: miR-24 Inhibits cell proliferation by targeting E2F2, MYC
and other cell-cycle genes via binding to 'seedless' 3′UTR microRNA
recognition elements. Mol Cell. 35:610–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lerner M, Lundgren J, Akhoondi S, Jahn A,
Ng HF, Akbari Moqadam F, Oude Vrielink JA, Agami R, Den Boer ML,
Grandér D and Sangfelt O: MiRNA-27a controls FBW7/hCDC4-dependent
cyclin E degradation and cell cycle progression. Cell Cycle.
10:2172–2183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ebert PJ, Jiang S, Xie J, Li QJ and Davis
MM: An endogenous positively selecting peptide enhances mature T
cell responses and becomes an autoantigen in the absence of
microRNA miR-181a. Nat Immunol. 10:1162–1169. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ventura A, Young AG, Winslow MM, Lintault
L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone
JR, et al: Targeted deletion reveals essential and overlapping
functions of the miR-17 through 92 family of miRNA clusters. Cell.
132:875–886. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Curtale G, Citarella F, Carissimi C,
Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V
and Macino G: An emerging player in the adaptive immune response:
MicroRNA-146a is a modulator of IL-2 expression and
activation-induced cell death in T lyzmphocytes. Blood.
115:265–273. 2010. View Article : Google Scholar
|
|
19
|
Yu Z, Ni L, Chen D, et al: Identification
of miR-7 as an oncogene in renal cell carcinoma. J Mol Histol.
44:669–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng H, Luo J, Hao H, Hu J, Xie SK, Ren D
and Rao B: MicroRNA-100 regulates SW620 colorectal cancer cell
proliferation and invasion by targeting RAP1B. Oncol Rep.
31:2055–2062. 2014.PubMed/NCBI
|
|
21
|
Xie J, Chen M, Zhou J, Mo MS, Zhu LH, Liu
YP, Gui QJ, Zhang L and Li GQ: miR-7 inhibits the invasion and
metastasis of gastric cancer cells by suppressing epidermal growth
factor receptor expression. Oncol Rep. 31:1715–1722.
2014.PubMed/NCBI
|
|
22
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Babashah S and Soleimani M: The oncogenic
and tumour suppressive roles of microRNAs in cancer and apoptosis.
Eur J Cancer. 47:1127–1137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Seca H, Almeida GM, Guimarães JE and
Vasconcelos MH: miR signatures and the role of miRs in acute
myeloid leukaemia. Eur J Cancer. 46:1520–1527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schickel R, Boyerinas B, Park SM and Peter
ME: MicroRNAs: Key players in the immune system, differentiation,
tumorigenesis and cell death. Oncogene. 27:5959–5974. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Müller S and Nowak K: Exploring the
miRNA-mRNA regulatory network in clear cell renal cell carcinomas
by next-generation sequencing expression profiles. Biomed Res Int.
2014:9484082014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Osanto S, Qin Y, Buermans HP, Berkers J,
Lerut E, Goeman JJ and van Poppel H: Genome-wide microRNA
expression analysis of clear cell renal cell carcinoma by next
generation deep sequencing. PLoS One. 7:e382982012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y,
Zhao X, Liang C, Wang Y, Sun L, et al: Integrated profiling of
microRNAs and mRNAs: MicroRNAs located on Xq27.3 associate with
clear cell renal cell carcinoma. PLoS One. 5:e152242010. View Article : Google Scholar
|
|
30
|
Edge SB and Compton CC: The American Joint
Committee on Cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Su Z, Chen D, Zhang E, et al:
MicroRNA-509-3p inhibits cancer cell proliferation and migration by
targeting the mitogen-activated protein kinase kinase kinase 8
oncogene in renal cell carcinoma. Mol Med Rep. 12:1535–1543.
2015.PubMed/NCBI
|
|
33
|
Patel C, Ahmed A and Ellsworth P: Renal
cell carcinoma: A reappraisal. Urol Nurs. 32:182–190.
2012.PubMed/NCBI
|
|
34
|
Yang SF, Hsu HL, Chao TK, Hsiao CJ, Lin YF
and Cheng CW: Annexin A2 in renal cell carcinoma: Expression,
function and prognostic significance. Urol Oncol. 33:e11–e21. 2015.
View Article : Google Scholar
|
|
35
|
Lang Y, Xu S, Ma J, Wu J, Jin S, Cao S and
Yu Y: MicroRNA-429 induces tumorigenesis of human non-small cell
lung cancer cells and targets multiple tumor suppressor genes.
Biochem Biophys Res Commun. 450:154–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang Y, Dai Y, Yang J, Chen T, Yin Y,
Tang M, Hu C and Zhang L: Microarray analysis of microRNA
expression in renal clear cell carcinoma. Eur J Surg Oncol.
35:1119–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Luthra R, Chen H, Roy-Chowdhuri S and
Singh RR: Next-generation sequencing in clinical molecular
diagnostics of cancer: Advantages and challenges. Cancers (Basel).
7. pp. 2023–2036. 2015, View Article : Google Scholar
|
|
38
|
Christensen LL, Tobiasen H, Holm A,
Schepeler T, Ostenfeld MS, Thorsen K, Rasmussen MH,
Birkenkamp-Demtroeder K, Sieber OM, Gibbs P, et al: MiRNA-362-3p
induces cell cycle arrest through targeting of E2F1, USF2 and PTPN1
and is associated with recurrence of colorectal cancer. Int J
Cancer. 133:67–78. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Van Wynsberghe PM, Chan SP, Slack FJ and
Pasquinelli AE: Analysis of microRNA expression and function.
Methods Cell Biol. 106:219–252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ishitani T and Ishitani S: Nemo-like
kinase, a multifaceted cell signaling regulator. Cell Signal.
25:190–197. 2013. View Article : Google Scholar
|
|
41
|
Yuzugullu H, Benhaj K, Ozturk N, Senturk
S, Celik E, Toylu A, Tasdemir N, Yilmaz M, Erdal E, Akcali KC, et
al: Canonical wnt signaling is antagonized by noncanonical wnt5a in
hepato-cellular carcinoma cells. Mol Cancer. 8:902009. View Article : Google Scholar
|
|
42
|
Stevens KN, Kelemen LE, Wang X, Fridley
BL, Vierkant RA, Fredericksen Z, Armasu SM, Tsai YY, Berchuck A,
Narod SA, et al: Common variation in nemo-like kinase is associated
with risk of ovarian cancer. Cancer Epidemiol Biomarkers Prev.
21:523–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yasuda J, Tsuchiya A, Yamada T, Sakamoto
M, Sekiya T and Hirohashi S: Nemo-like kinase induces apoptosis in
DLD-1 human colon cancer cells. Biochem Biophys Res Commun.
308:227–233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Emami KH, Brown LG, Pitts TE, Sun X,
Vessella RL and Corey E: Nemo-like kinase induces apoptosis and
inhibits androgen receptor signaling in prostate cancer cells.
Prostate. 69:1481–1492. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jung KH, Kim JK, Noh JH, Eun JW, Bae HJ,
Xie HJ, Ahn YM, Park WS, Lee JY and Nam SW: Targeted disruption of
Nemo-like kinase inhibits tumor cell growth by simultaneous
suppression of cyclin D1 and CDK2 in human hepatocellular
carcinoma. J Cell Biochem. 110:687–696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cui G, Li Z, Shao B, Zhao L, Zhou Y, Lu T,
Wang J, Shi X, Wang J, Zuo G, et al: Clinical and biological
significance of nemo-like kinase expression in glioma. J Clin
Neurosci. 18:271–275. 2011. View Article : Google Scholar
|