Introduction
Ulcerative colitis and Crohn's disease (CD) are two
major types of inflammatory bowel disease (IBD), which influence
millions of individuals (1,2);
however, the pathogenesis and etiology of IBD remain to be
elucidated. Pathological alterations associated with IBD include
gut inflammation, disruption of the intestinal epithelial barrier
and ulcer formation (1,2). The intestinal epithelial barrier
consists of a monolayer of epithelial cells with intercellular
tight junctions, which regulates gut permeability (3). Excessive apoptosis of intestinal
epithelial cells (IECs) may compromise the intestinal barrier
function, and has been recognized as a major pathogenic mechanism
in the process of chronic intestinal inflammation (3).
Angiotensin II is a proinflammatory peptide that is
associated with digestive system disorders, particularly
inflammation (4–7). Previous studies have demonstrated
that colonic mucosal levels of angiotensin I and II are
significantly elevated in patients with CD (4,5). In
addition, amelioration of 2,4,6-trinitrobenzenesulphonic acid
(TNBS)-induced colitis was observed in angiotensinogen knockout
mice (8), and dextran sulfate
sodium (DSS)-induced colonic inflammation was reduced in
angiotensin II type 1 receptor (AT1R)-deficient mice (9). However, little is currently known
regarding the possible pathological mechanism of the angiotensin
II-AT1R pathway in intestinal inflammation.
Angiotensin II has previously been demonstrated to
induce apoptosis via AT1R in myocytes and IECs, and the underlying
mechanism may be partially due to its regulatory effect on the
B-cell lymphoma 2 (Bcl-2)/Bcl-2-associated X protein (Bax) pathway
(10,11). Bax and Bcl-2 are important
proapoptotic and anti-apoptotic mediators, respectively, which have
a crucial role in regulating IEC apoptosis via the intrinsic
apoptotic pathway (12–14). The ratio of Bcl-2 to Bax is
frequently used as an indicator of survival potential, in which a
high ratio protects against apoptosis and a low ratio promotes
apoptosis (12–14).
Although the AT1R blocker losartan has been reported
to inhibit the apoptosis of IECs in vitro (11), its effects in vivo, as well
as the underlying mechanism, remain to be clarified. The present
study investigated the influence of losartan treatment on
TNBS-induced colitis, which is a common mouse model of CD (15). It was hypothesized that losartan
could attenuate TNBS-induced colitis by inhibiting the apoptosis of
IECs, through upregulating the ratio of Bcl-2 to Bax.
Materials and methods
Mouse model of TNBS-induced colitis
Male adult C57BL/6 J mice, (8–12 weeks old; 20–25
g), were purchased from the Center for Experimental Animals of
China Medical University (Shenyang, China). The animals were housed
in specific-pathogen free conditions. Mice were provided with food
and water ad libitum and maintained in a 12 h dark/light
cycle at 25°C. All animal procedures were reviewed and approved by
the Institutional Ethical Committee of China Medical University.
Mice were anesthetized by an intra-peritoneal injection with a
cocktail of xylazine (Rompun 2%; Bayer AG, Leverkusen, Germany) and
ketamine (Ketavest; 100 mg/ml; Pfizer, Inc., New York, NY, USA).
TNBS was prepared by dissolving 5% TNBS (Sigma-Aldrich, St. Louis,
MO, USA) in an equal volume of 100% ethanol, in order to generate a
working solution of 2.5% TNBS in 50% ethanol. To induce colitis,
the mice were administered 100 mg/kg TNBS into the rectum using an
18-gauge stainless steel gavage needle. The vehicle group was given
the same volume of 50% ethanol.
Losartan treatment
Losartan (Cozaar, Merck & Co., Whitehouse
Station, NJ, USA) was administered orally in distilled drinking
water (~10 mg/kg/day) for 2 weeks prior to the induction of
colitis, and after TNBS administration until the time of sacrifice.
This dose was similar to that used in previous studies regarding
this drug (16). The vehicle group
was given distilled drinking water only.
Western blotting
The mice were sacrificed by cervical dislocation
following anesthetization with carbon dioxide, on day 2 after TNBS
treatment. The colon was cut open and washed with
phosphate-buffered saline, following which the colonic mucosa was
harvested and homogenized in radioimmunopre-cipitation assay buffer
(Beyotime Institute of Biotechnology, Haimen, China). The
supernatant was used for the measurement of protein concentration
using a bicinchoninic acid assay (Beyotime Institute of
Biotechnology). Subsequently, 3X SDS was added and the mixture was
heated to 95°C for 5 min. The protein lysates (50 µg/lane)
were separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, and the proteins were electrophoretically
transferred onto polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). The membranes were blocked with 5%
non-fat milk at room temperature for 1 h, and then washed three
times in 0.1% Tris-buffered saline with Tween 30, with agitation
for 10 min. The membranes were then incubated with primary and
secondary antibodies. The blots were visualized using an enhanced
chemiluminescence kit (Santa Cruz Biotechnology, Inc.) and ImageJ
software (1.47v; National Institutes of Health, Bethesda, MD, USA)
was used to measure the density of the bands, which were normalized
to β-actin. The antibodies used in the present study included:
Mouse monoclonal anti-β-actin (cat. no. A1978; 1:10,000;
Sigma-Aldrich), rabbit polyclonal anti-Bcl-2 (cat. no. 2876, Cell
Signaling Technology, Inc., Beverly, MA, USA; 1:2,000), rabbit
polyclonal anti-Bax (cat. no. 2772; 1:2,000; Cell Signaling
Technology, Inc.) and rabbit polyclonal anti-caspase 3 (cat. no.
9662; 1:1,000; Cell Signaling Technology, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Mice were sacrificed on day 2 after TNBS treatment.
A section of distal colon, ~1.0 cm in length, was harvested from
the same segment in all of the mice and RNA was isolated from the
colonic mucosa using TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). First-strand cDNA was
synthesized from 2 µg total RNA in a 20 µl reaction
system using M-MLV reverse transcriptase (Thermo Fisher Scientific,
Inc.) and random primers (Takara Bio, Inc., Otsu, Japan). qPCR was
performed using a Roche LightCycler Real Time PCR system with SYBR
green PCR Master mix (Takara Bio, Inc., Otsu, Japan). Relative mRNA
transcription levels were calculated according to the
2−ΔΔCq formula. β-2 microglobulin was used as an
internal control. PCR primer sequences are presented in Table I. The PCR reaction conditions were
as follows: 95°C for 2 min, followed by 40 cycles of 95°C for 15
sec, 55°C for 15 sec, 72°C for 1 min, 72°C for 5 min and then kept
at 4°C until tubes were removed.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Primer name | Forward (5′-3′) | Reverse (3′-5′) |
|---|
| TNF-α |
ATGAGCACAGAAAGCATGA |
AGTAGACAGAAGAGCGTGGT |
| IFN-γ |
TTCTTCAGCAACAGCAAGGC |
TCAGCAGCGACTCCTTTTCC |
| IL-1β |
AATGAAAGACGGCACACCCA |
TGCTTGTGAGGTGCTGATGT |
| IL-2 |
TTGTGCTCCTTGTCAACAGC |
CTGGGGAGTTTCAGGTTCCT |
| IL-6 |
CCTCTGGTCTTCTGGAGTACC |
ACTCCTTCTGTGACTCCAGC |
| IL-12 |
GCACCAAATTACTCCGGACG |
TGGTCCAGTGTGACCTTCTC |
| IL-17 |
TCTCCACCGCAATGAAGACC |
CACACCCACCAGCATCTTCT |
| IL-23 |
GCTGTGCCTAGGAGTAGCAG |
TGGCTGTTGTCCTTGAGTC |
| β-2
microglobulin |
CGGCCTGTATGCTATCCAGA |
GGGTGAATTCAGTGTGAGCC |
Hematoxylin & eosin staining
The mice were sacrificed 4 days after TNBS
treatment, and images of the colonic morphology were captured using
a Sony digital camera Sony digital camera (DSC-TX9C; Sony
Corporation, Tokyo, Japan). The damage was scored according to a
macroscopic scoring system (17).
The distal colon was harvested and fixed overnight with 4%
formaldehyde, dehydrated with graded alcohol, placed in xylene and
embedded in paraffin. Sections (4 µm) were stained with
hematoxylin & eosin (Beyotime Institute of Biotechnology). Five
areas were randomly selected in each section and examined at ×100
magnification. In each field, colon microscopic scoring (Leica
DFC425; Leica Microsystems GmbH, Wetzlar, Germany) was performed by
two pathologists independently that were blind to the study design,
according to the following microscopic scoring system (17): Loss of mucosal architecture,
cellular infiltration and muscle thickening were scored 0,1,2 or 3
(absent, mild-severe); crypt abscess formation and goblet cell
depletion were scored 0 or 1 (absent or present).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) staining
Apoptotic cells in the colonic sections were
identified according to the TUNEL staining method using a
commercial kit (Roche Diagnostics Corp., Indianapolis, IN, USA),
according to the manufacturer's protocol. Apoptotic index was
defined as the percentage of TUNEL-positive-cells (red staining) in
100 randomly chosen IECs, as previously described (18).
Measurement of intestinal
permeability
Mice were denied access to food, but were allowed
water for 4 h prior to gavage. At 45 h post-TNBS injection, 50
mg/ml fluorescein isothio-cyanate-4 kD dextran (FD4; Sigma-Aldrich)
was gavaged at 4 µl/g body weight through an 18-guage
stainless steel gavage needle. After 3 h (48 h post-TNBS
injection), the mice were anesthetized via intraperitoneal
injection with a cocktail of xylazine and ketamine. Blood was
collected from the inferior vena cava with a 1 ml syringe and
rested overnight in 4°C. The blood was then centrifuged at 10,000 x
g for 10 min at 4°C, the supernatant was collected and blood serum
was harvested, of which 200 µl was added to each well of a
96-well plate. Subsequently, the serum concentration of FD4 was
measured using a Synergy HT plate reader (BioTek Laboratories,
Inc., Winooski, VT, USA), as previously described (19).
Statistical analysis
All continuous data are presented as the mean ±
standard deviation. Statistical comparisons of continuous variables
between groups were conducted using Student's t-test or one-way
analysis of variance. Graphpad Prism software 6.0 (Graphpad
Software Inc., La Jolla, CA, USA) and SPSS 17.0 (SPSS, Inc.,
Chicago, IL, USA) were used to perform data analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of losartan on TNBS-induced
colitis
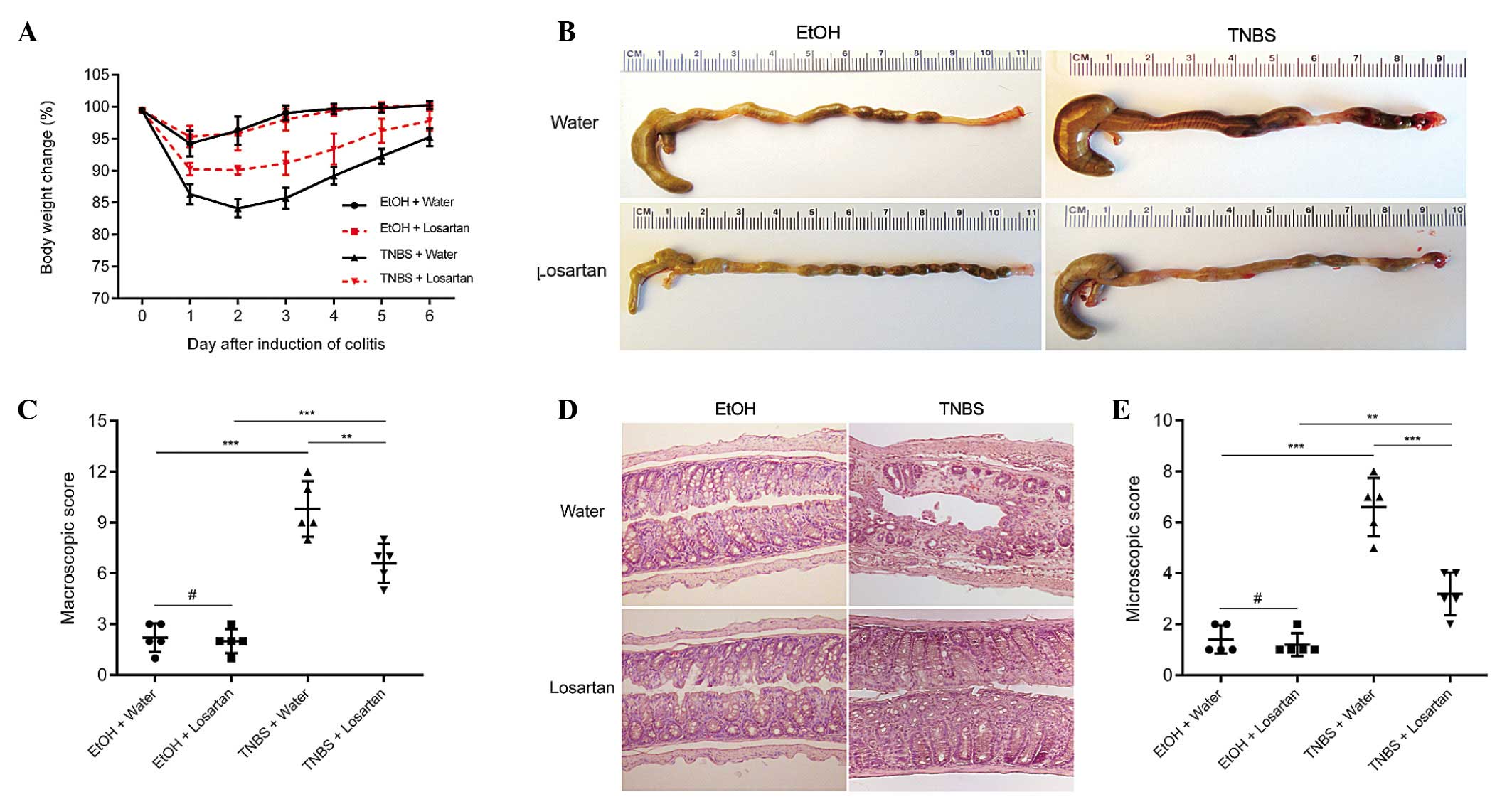
TNBS successfully induced colitis, as evidenced by
marked weight loss, hyperemia, inflammation and ulcerative necrosis
in the colon tissue of the TNBS-treated mice, as compared with the
ethanol-treated control group, which exhibited only negligible
pathological alterations. TNBS-induced weight loss was ameliorated
and the colonic morphology was partly restored following treatment
with losartan (Fig. 1A–C).
Microscopically, transmural inflammation characterized by
infiltration of inflammatory cells, mucosal and submucosal
ulcerations, loss of goblet cells and fibrosis was observed
throughout the whole colon in the TNBS-treated mice. Losartan
largely rescued the pathological alterations and restored the
normal histological structure of the colonic mucosa and submucosa
(Fig. 1D and E). These results
suggest that losartan may exert protective effects against
TNBS-induced colitis.
Losartan suppresses proinflammatory
cytokine expression in colonic mucosa
Necrotic inflammation was observed in the colonic
mucosa and submucosa of the TNBS-treated mice, which could be
partly inhibited by losartan. To further explore the mechanism
underlying this effect, the relative expression levels of
proinflammatory cytokines were detected. Since TNBS induces colitis
via a Th1-mediated response, production of the following Th1
cytokines was examined: Tumor necrosis factor (TNF)-α, interferon
(IFN)-γ, interleukin (IL)-2, IL-12, IL-1β and IL-6 (20). In addition, IL-17 and IL-23, which
are derived from Th-17 inflammatory cells, were demonstrated to
participate in the inflammatory process of TNBS-induced colitis
(20). All of the previously
mentioned cytokines were increased following stimulation with TNBS.
Losartan was able to significantly ameliorate this tendency for all
Th1-mediated cytokines, and was able to decrease the expression
levels of the Th17-mediated cytokines but not significantly
(Fig. 2).
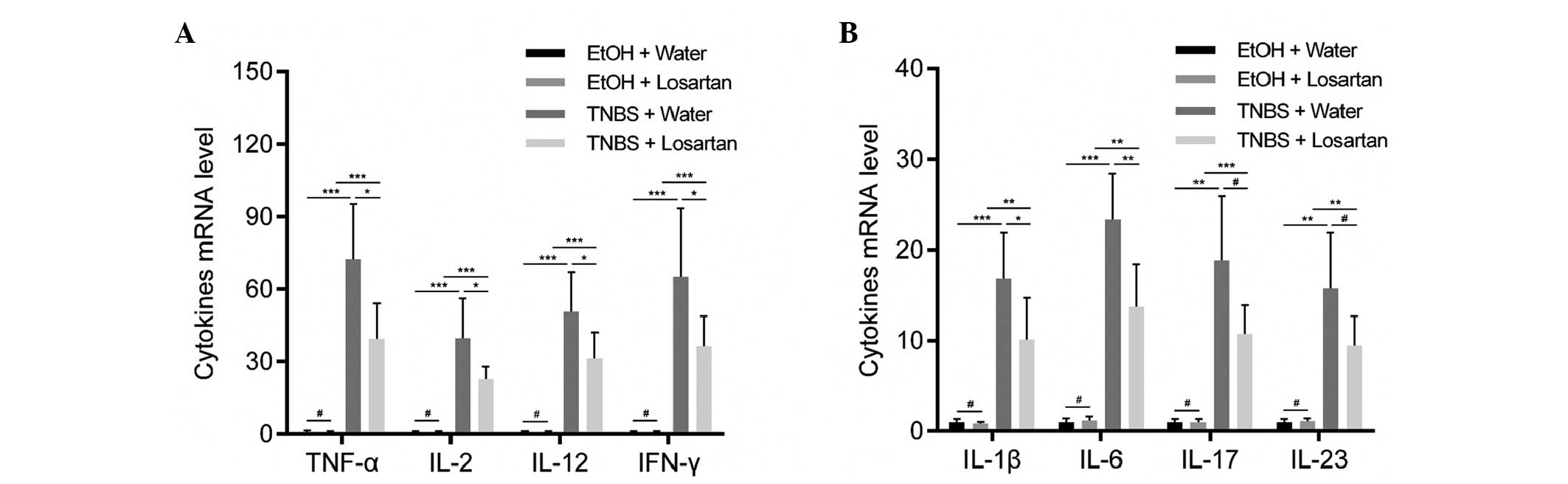 | Figure 2Losartan suppressed
2,4,6-trinitrobenzenesulphonic acid (TNBS)-induced colonic
inflammation. Reverse transcription-quantitative polymerase chain
reaction was conducted to detect the expression levels of the
following proinflammatory cytokines: (A) Tumor necrosis factor
(TNF-α), interleukin (IL)-2, IL-12, interferon (IFN)-γ, and (B)
IL-1β, IL-6, IL-17 and IL-23 in the colonic mucosa from each
treatment group on day 2 (n=6/group). #P>0.05,
*P<0.05, **P<0.01,
***P<0.001. EtOH, ethanol. |
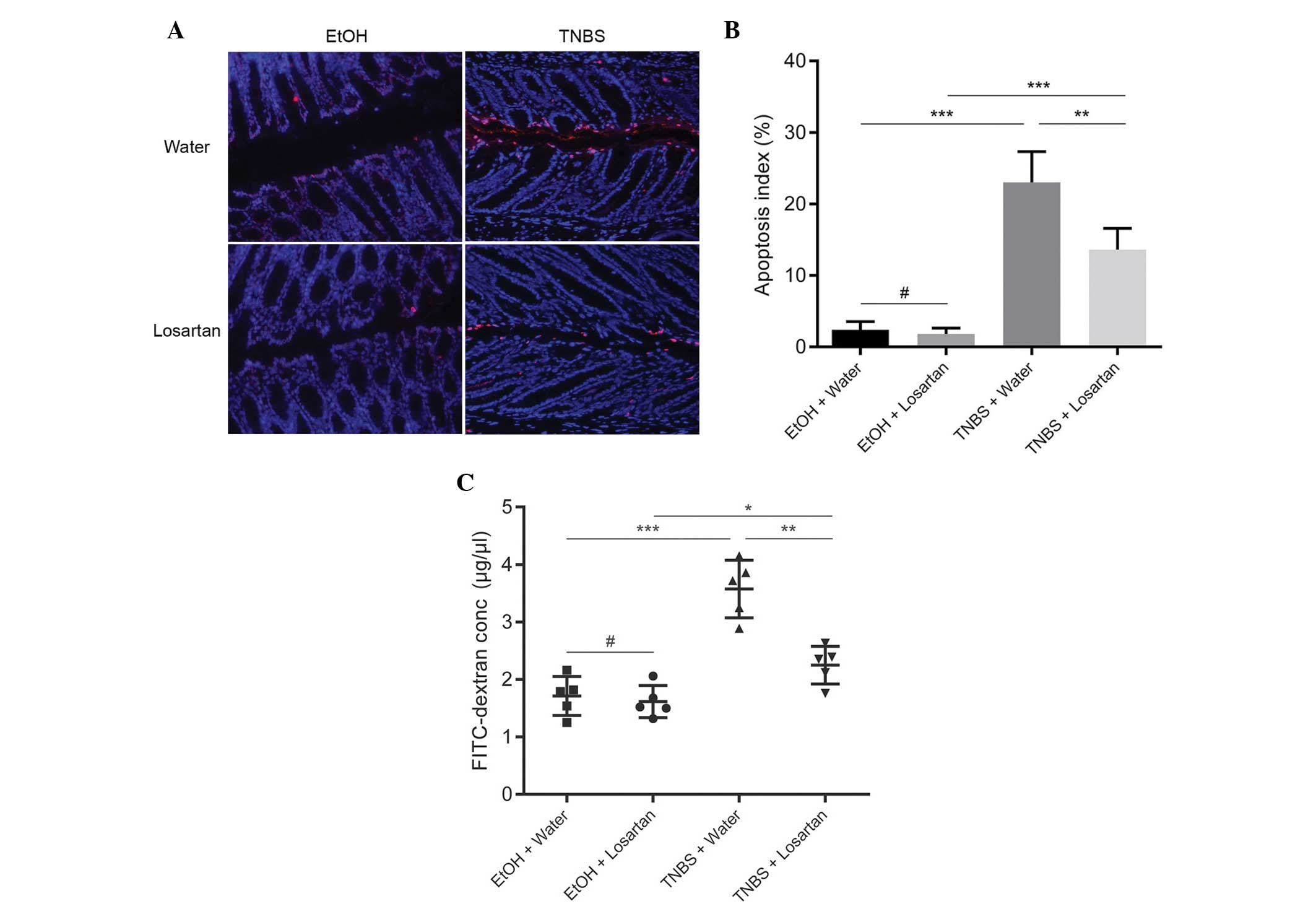
Losartan decreases intestinal
permeability by inhibiting IEC apoptosis
Since losartan had been demonstrated to have an
anti-apoptotic effect in vitro (11), the present study investigated
whether the protective effect of losartan on TNBS-induced colitis
was associated with its anti-apoptotic effect on IECs in
vivo. IEC apoptosis may lead to disrupted intestinal barrier
function and increased gut permeability, as reflected by increased
leakage of FD4 from the lumen into the circulation. An abundance of
apoptotic IECs were observed following TNBS administration, whereas
the number of apoptotic epithelial cells was markedly reduced in
the TNBS + Losartan mice (Fig. 3A and
B). Concordantly, treatment with losartan partly alleviated the
FD4 leak by decreasing intestinal permeability (Fig. 3C). These results indicate that
losartan may preserve the intestinal epithelial barrier and
decrease gut permeability by inhibiting the apoptosis of IECs.
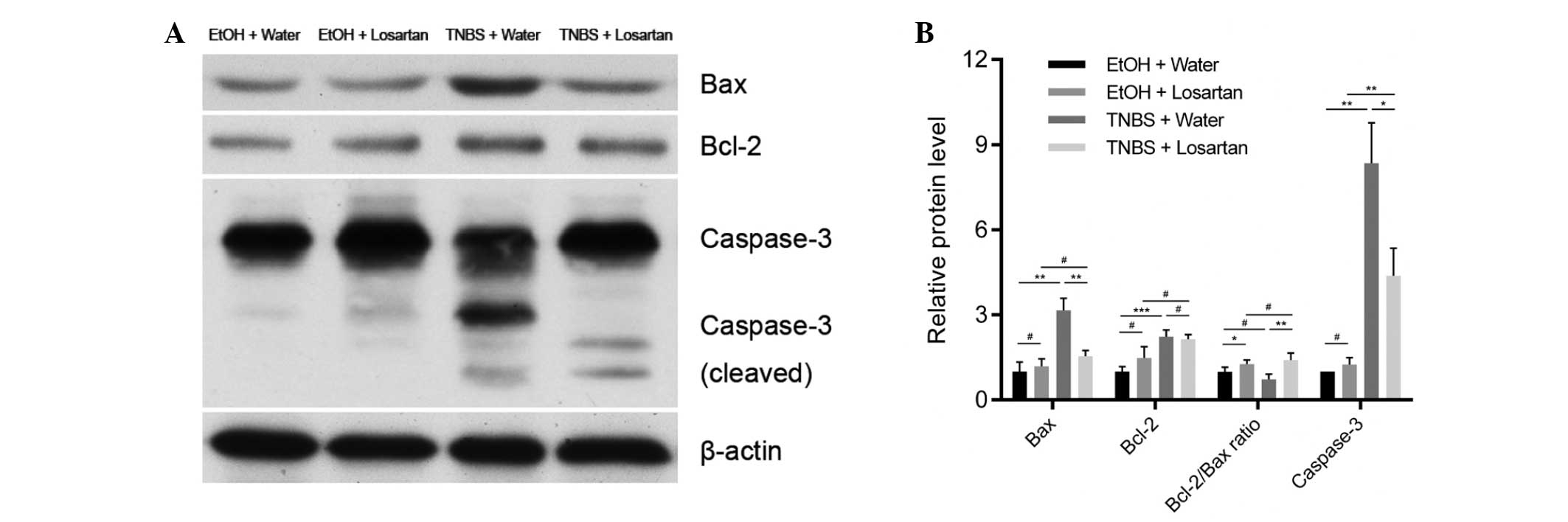
Losartan attenuates caspase-3 mediated
apoptosis through upregulating the Bcl-2/Bax ratio
The mechanism underlying the anti-apoptotic effect
of losartan on IECs was further investigated. Following treatment
with TNBS, the expression levels of the proapoptotic protein Bax
were markedly increased, whereas the expression levels of the
anti-apoptotic protein Bcl-2 were slightly elevated. There was a
significant decrease in the protein expression levels of Bax, but
not of Bcl-2, in the TNBS + Losartan group, as compared with the
TNBS + Water group (Fig. 4A and
B), which suggests that losartan treatment may increase the
Bcl-2/Bax ratio. Concordantly, the expression levels of cleaved
caspase-3 were markedly inhibited in the TNBS + Losartan group.
These data indicate that the anti-apoptotic mechanism of losartan
was, at least in part, attributed to the inhibition of caspase-3
induction through upregulation of the Bcl-2/Bax ratio.
Discussion
The results of the present study revealed the
protective role of the AT1R blocker losartan in TNBS-induced
colitis. Losartan was able to relieve the symptoms of TNBS-induced
colitis and ameliorate the induction of proinflammatory cytokine
expression and IEC apoptosis. Its inhibitory effects on IEC
apoptosis were partially mediated through increasing the Bcl-2/Bax
ratio and subsequently inhibiting the induction of caspase-3. These
observations provided a novel insight into the pathogenesis of
TNBS-induced colitis, which serves as a model of CD (15). These results suggest that AT1R
blockers may be a potential therapeutic option for patients with
IBD.
The classical renin-angiotensin system (RAS)
maintains body fluid and electrolyte homeostasis, and a previous
study disclosed the link between RAS and IBD (21). Angiotensin II, which is the major
biologically active component of the RAS, is involved in apoptosis,
vascular remodeling and inflammation (21). Jaszewski et al (4) reported that levels of angiotensin I
and II were higher in patients with CD, and this was correlated
with the degree of inflammation. Therefore the present study
inhibited this pathway using the AT1R blocker losartan, in order to
determine whether colonic inflammation could be relieved. Mice
treated with losartan exhibited a less severe inflammatory
response, as compared with those treated with water only after the
administration of TNBS. In addition, the elevation of
proinflammatory cytokines was reduced following treatment with
losartan.
RAS has an important role in intestinal inflammation
in vitro and in vivo. A previous study demonstrated
that losartan was able to significantly attenuate angiotensin
II-induced IEC apoptosis in cultured HT-29 cell lines (11). Angiotensin converting enzyme
inhibitors have been demonstrated to markedly attenuate colon
inflammation in animal models of colitis (21,22).
Furthermore, angiotensinogen gene knockout mice exhibited reduced
colitis, as compared with wild type mice following TNBS treatment
(8). AT1R-deficient mice also
exhibited mild DSS-induced acute colonic inflammation (9).
Angiotensin II has been shown to promote apoptosis
through AT1R in numerous circumstances (10,11),
including DSS-induced colitis in mice (23). Therefore, preservation of the
intestinal epithelial barrier may be due to the anti-apoptotic
effect of losartan via blockade of the Angiotensin II-AT1R
signaling pathway. Treatment with AT1R blockers has previously been
reported to attenuate bleomycin-induced pulmonary epithelial
apoptosis in mice (24). In the
present study, mice treated with losartan displayed reduced colonic
damage following TNBS administration. In addition, the expression
levels of caspase-3, a downstream apoptotic protein, were
significantly reduced by losartan. These results indicated that
losartan was able to attenuate angiotensin II-mediated apoptosis
and thus relieve the symptoms of TNBS-induced colitis.
Apoptosis is regulated by a complex interplay
between proapoptotic and anti-apoptotic mediators, including Bax
and Bcl-2 (12–14). A previous study suggested that
angiotensin II-induced IEC apoptosis was associated with the
Bcl-2/Bax intrinsic pathway (11).
At present, the Bcl-2 protein family is known to consist of 25
homogenous members, including proapoptotic proteins, such as
Bcl-2-associated death promoter, BH3 interacting-domain death
agonist and Bax, as well as anti-apoptotic proteins such as Bcl-2,
Bcl-extra large and Bcl-2-like protein 2 (14). Bcl-2 suppresses apoptosis by
inhibiting the release of cytochrome c from the mitochondria
into the cytoplasm (14). The
ratio of Bcl-2/Bax is frequently used as an indicator of survival
potential, in which a high ratio protects against apoptosis and a
low ratio favors apoptosis (12–14).
The present study demonstrated that the Bcl-2/Bax ratio was
markedly upregulated by losartan, which coincided with a decrease
in caspase-3 induction. Regulation of the Bcl-2/Bax intrinsic
pathway may contribute to the mechanism underlying the effects of
the AT1R pathway on alleviating IEC apoptosis.
Angiotensin II receptors are expressed not only in
colonic mucosa (25) but also in
microvascular endothelial cells (26). The microvascular system also has an
important role in colonic inflammation. Changes in microvascular
circulation have been associated with colitis in previous animal
studies (26,27), however, colonic blood flow has been
reported to be reduced in several colitis models (27,28,29).
In the present study, the curative effects of losartan may in part
be attributed to its benefits on the microcirculation; however,
this requires further investigation.
In conclusion, the present study demonstrated that
the AT1R blocker losartan was able to inhibit the apoptosis of
IECs, and therefore attenuate TNBS-induced colitis. The effects of
losartan may have been mediated, at least in part, through
increasing the ratio of Bcl-2/Bax and subsequently suppressing the
expression of the proapoptotic mediator caspase-3. Given the active
RAS status observed in patients with IBD, AT1R blocker may be a
potential therapeutic agent for the treatment of IBD.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81271938), Liaoning
Science and Technology Project (grant no. 2013225079) and the
Outstanding Scientific Fund of Shengjing Hospital.
References
|
1
|
Abraham C and Cho JH: Inflammatory bowel
disease. N Engl J Med. 361:2066–2078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Podolsky DK: Inflammatory bowel disease. N
Engl J Med. 347:417–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Peterson LW and Artis D: Intestinal
epithelial cells: Regulators of barrier function and immune
homeostasis. Nat Rev Immunol. 14:141–153. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jaszewski R, Tolia V, Ehrinpreis MN,
Bodzin JH, Peleman RR, Korlipara R and Weinstock JV: Increased
colonic mucosal angiotensin I and II concentrations in Crohn's
colitis. Gastroenterology. 98:1543–1548. 1990.PubMed/NCBI
|
|
5
|
Garg M, Burrell LM, Velkoska E, Griggs K,
Angus PW, Gibson PR and Lubel JS: Upregulation of circulating
components of the alternative renin-angiotensin system in
inflammatory bowel disease: A pilot study. J Renin Angiotensin
Aldosterone Syst. 16:559–569. 2015. View Article : Google Scholar
|
|
6
|
Bregonzio C, Armando I, Ando H, Jezova M,
Baiardi G and Saavedra JM: Anti-inflammatory effects of angiotensin
II AT1 receptor antagonism prevent stress-induced gastric injury.
Am J Physiol Gastrointest Liver Physiol. 285:G414–G423. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kuno A, Yamada T, Masuda K, Ogawa K,
Sogawa M, Nakamura S, Nakazawa T, Ohara H, Nomura T, Joh T, et al:
Angiotensin-converting enzyme inhibitor attenuates pancreatic
inflammation and fibrosis in male Wistar Bonn/Kobori rats.
Gastroenterology. 124:1010–1019. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inokuchi Y, Morohashi T, Kawana I,
Nagashima Y, Kihara M and Umemura S: Amelioration of
2,4,6-trinitrobenzene sulphonic acid induced colitis in
angiotensinogen gene knockout mice. Gut. 54:349–356. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Katada K, Yoshida N, Suzuki T, Okuda T,
Mizushima K, Takagi T, Ichikawa H, Naito Y, Cepinskas G and
Yoshikawa T: Dextran sulfate sodium-induced acute colonic
inflammation in angiotensin II type 1a receptor deficient mice.
Inflamm Res. 57:84–91. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leri A, Claudio PP, Li Q, Wang X, Reiss K,
Wang S, Malhotra A, Kajstura J and Anversa P: Stretch-mediated
release of angiotensin II induces myocyte apoptosis by activating
p53 that enhances the local renin-angiotensin system and decreases
the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest.
101:1326–1342. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang W, Sun L, Xiao W and Yang H:
Essential role of angiotensin receptors in the modulation of
intestinal epithelial cell apoptosis. J Pediatr Gastroenterol Nutr.
57:562–569. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pawlowski J and Kraft AS: Bax-induced
apoptotic cell death. Proc Natl Acad Sci USA. 97:529–531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tang Y, Swartz-Basile DA, Swietlicki EA,
Yi L, Rubin DC and Levin MS: Bax is required for resection-induced
changes in apoptosis, proliferation, and members of the extrinsic
cell death pathways. Gastroenterology. 126:220–230. 2004.
View Article : Google Scholar
|
|
14
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View
Article : Google Scholar
|
|
15
|
Morris GP, Beck PL, Herridge MS, Depew WT,
Szewczuk MR and Wallace JL: Hapten-induced model of chronic
inflammation and ulceration in the rat colon. Gastroenterology.
96:795–803. 1989.PubMed/NCBI
|
|
16
|
Wengrower D, Zanninelli G, Latella G,
Necozione S, Metanes I, Israeli E, Lysy J, Pines M, Papo O and
Goldin E: Losartan reduces trinitrobenzene sulphonic acid-induced
colorectal fibrosis in rats. Can J Gastroenterol. 26:33–39.
2012.PubMed/NCBI
|
|
17
|
Appleyard CB and Wallace JL: Reactivation
of hapten-induced colitis and its prevention by anti-inflammatory
drugs. Am J Physiol. 269:G119–G125. 1995.PubMed/NCBI
|
|
18
|
Liu W, Chen Y, Golan MA, Annunziata ML, Du
J, Dougherty U, Kong J, Musch M, Huang Y, Pekow J, et al:
Intestinal epithelial vitamin D receptor signaling inhibits
experimental colitis. J Clin Invest. 123:3983–3996. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Su L, Nalle SC, Shen L, Turner ES, Singh
G, Breskin LA, Khramtsova EA, Khramtsova G, Tsai PY, Fu YX, et al:
TNFR2 activates MLCK-dependent tight junction dysregulation to
cause apoptosis-mediated barrier loss and experimental colitis.
Gastroenterology. 145:407–415. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brand S: Crohn's disease: Th1, Th17 or
both? The change of a paradigm: New immunological and genetic
insights implicate Th17 cells in the pathogenesis of Crohn's
disease. Gut. 58:1152–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hume GE and Radford-Smith GL: ACE
inhibitors and angiotensin II receptor antagonists in Crohn's
disease management. Expert Rev Gastroenterol Hepatol. 2:645–651.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spencer AU, Yang H, Haxhija EQ, Wildhaber
BE, Greenson JK and Teitelbaum DH: Reduced severity of a mouse
colitis model with angiotensin converting enzyme inhibition. Dig
Dis Sci. 52:1060–1070. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Okawada M, Koga H, Larsen SD, Showalter
HD, Turbiak AJ, Jin X, Lucas PC, Lipka E, Hillfinger J, Kim JS and
Teitelbaum DH: Use of enterally delivered angiotensin II type 1a
receptor antagonists to reduce the severity of colitis. Dig Dis
Sci. 56:2553–2565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Rayford H and Uhal BD: Essential
roles for angiotensin receptor AT1a in bleomycin-induced apoptosis
and lung fibrosis in mice. Am J Pathol. 163:2523–2530. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hirasawa K, Sato Y, Hosoda Y, Yamamoto T
and Hanai H: Immunohistochemical localization of angiotensin II
receptor and local renin-angiotensin system in human colonic
mucosa. J Histochem Cytochem. 50:275–282. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Petnehazy T, Cooper D, Stokes KY, Russell
J, Wood KC and Granger DN: Angiotensin II type 1 receptors and the
intestinal microvascular dysfunction induced by ischemia and
reperfusion. Am J Physiol Gastrointest Liver Physiol.
290:G1203–G1210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mori M, Stokes KY, Vowinkel T, Watanabe N,
Elrod JW, Harris NR, Lefer DJ, Hibi T and Granger DN: Colonic blood
flow responses in experimental colitis: Time course and underlying
mechanisms. Am J Physiol Gastrointest Liver Physiol.
289:G1024–G1029. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deniz M, Cetinel S and Kurtel H: Blood
flow alterations in TNBS-induced colitis: Role of endothelin
receptors. Inflamm Res. 53:329–336. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garrelds IM, Heiligers JP, Van Meeteren
ME, Duncker DJ, Saxena PR, Meijssen MA and Zijlstra FJ: Intestinal
blood flow in murine colitis induced with dextran sulfate sodium.
Dig Dis Sci. 47:2231–2236. 2002. View Article : Google Scholar : PubMed/NCBI
|