Introduction
Diabetic cardiomyopathy (DCM) is characterized
clinically by diastolic dysfunction in the early stage, evolving to
systolic dysfunction in the final stage. Impaired ejection fraction
is a severe complication that affects a notable proportion of
diabetic patients (1). With the
rising incidence of diabetes, DCM is an increasingly important
disease in the cardiovascular field. Myocardial fibrosis results
from a disproportionate increase in collagen deposition in the
extracellular matrix (ECM). It is one of the predominant
pathological features of DCM (2)
and it is a critical determinant of the development of diastolic
and systolic dysfunction in patients with DCM (3). However, there are no effective
therapeutic strategies currently available that prevent the
progression of diabetic myocardial fibrosis. Further elucidation of
the mechanisms underlying myocardial fibrosis in diabetic patients
is required and potential therapeutic strategies to limit
myocardial remodeling in DCM remain of interest to reduce the risk
of progression to heart failure.
Hydrogen sulfide (H2S), which was
previously considered to be a highly toxic gas, has been identified
as a novel gaseous signaling molecule (4), and similar to carbon monoxide and
nitric oxide, it exerts diverse effects on multiple physiological
and pathological processes in various tissues and organs (5). Recently, research into H2S
in the pathogenesis of cardiovascular disorders has rapidly
increased as the roles of H2S in the cardiovascular
system, including vasorelaxation, inhibition of vascular
remodeling, anti-atherosclerosis and cardioprotection, have been
demonstrated (5–8). In the heart, endogenous
H2S is predominantly generated from L-cysteine by
cystathionine-γ-lyase (CSE) (9,10).
Previous studies have demonstrated that H2S ameliorates
myocardial ischemia-reperfusion injury by attenuating cardiomyocyte
apoptosis, preserving mitochondrial function (11,12)
and alleviating structural and functional deterioration of the left
ventricle in ischemia-induced heart failure by attenuating
oxidative stress (13).
Furthermore, a recent study indicated that the decreased generation
of endogenous H2S is important in the development of
diabetic nephropathy in diabetic rats and administration of
H2S may delay the progress of diabetic nephropathy by
reducing mesangial cell proliferation, decreasing inflammation and
oxidative stress, and inhibiting the activity of the
renin-angiotensin system (14). In
addition, it has been demonstrated that H2S improves
left ventricular function in smoking rats by reducing autophagy
(15). However, the effect of
H2S on diabetic myocardial fibrosis and its underlying
mechanisms remain to be elucidated.
Autophagy is a lysosome-dependent process, which
degrades dysfunctional organelles and damaged proteins to maintain
cellular homeostasis and structural integrity of the cell.
Autophagy represents an evolutionarily conserved process for bulk
degradation and recycling of cytoplasmic components (16–18).
However, prolonged activation of autophagy may be observed in the
heart under several cardiotoxic stressors, including hyperglycemia,
ischemia-reperfusion and chronic high pressure load, this
accelerates cell death and results in the occurrence of associated
cardiovascular disease (19,20).
In the present study, a streptozotocin (STZ)-induced
diabetic rat model was used to evaluate the effects of
H2S on diabetic myocardial fibrosis, and furthermore, to
investigate the effects of H2S on
phosphatidylinositol-4,5-bisphosphate 3-kinase/RAC-α
serine/threonine-protein kinase (PI3K/AKT1)-regulated autophagy in
order to elucidate its underlying mechanisms.
Materials and methods
Animals and reagents
The experimental protocol was approved by the Animal
Ethics Committee of the University of South China (Hengyang,
China). Adult male Sprague-Dawley rats (weight, 288–300 g) were
obtained from the SJA Lab Animal Center of Changsha (Changsha,
China). The rats were housed in separate cages and had free access
to food and water. Animals were kept in a climate-controlled room
with a 12-h light/dark cycle. Sodium hydrosulfide (NaHS) was
purchased from Sigma-Aldrich (St. Louis, MO, USA). STZ was
purchased from MP Biomedicals, LLC (Santa Ana, CA, USA). A
hydroxyproline detection kit was obtained from Nanjing Jiancheng
Bioengineering Institute (Nanjing, China). Rabbit polyclonal
antibody against CSE (cat. no. sc-135203; used at 1:1,000 dilution)
was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). Rabbit polyclonal anti-collagen I (cat. no. BA0235), rabbit
polyclonal anti-collagen III (cat. no. BA0326), rabbit polyclonal
anti-matrix metalloproteinase (MMP)7 (cat. no. BA2110), rabbit
polyclonal anti-MMP8 (cat. no. BA2201), rabbit polyclonal
anti-MMP14 (cat. no. BA1278), rabbit polyclonal anti-tissue
inhibitor of metalloproteinase 1 (TIMP1; cat. no. BA3727), rabbit
polyclonal anti-transforming growth factor β 1 (TGFβ1; cat. no.
BA0290), rabbit polyclonal anti-PI3K (cat. no. BA0351), rabbit
polyclonal anti-AKT1 (cat. no. BA0631) and rabbit polyclonal
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH; cat. no.
PB0141) were all purchased from Wuhan Boster Biological Technology,
Ltd. (Wuhan, China). The dilution rate of these antibodies was
1:400. Furthermore, rabbit monoclonal anti-Beclin1 (cat. no. 3495),
rabbit monoclonal anti-autophagy-related protein (Atg; cat. no.
3415), rabbit monoclonal anti-Atg5 (cat. no. 12994) and rabbit
monoclonal anti-Atg16 (cat. no. 8089) were purchased from Cell
Signaling Technology, Inc, (Danvers, MA, USA). The dilution rate of
these antibodies was 1:1,000. Anti-rabbit secondary antibody (cat.
no. 074–1506) was obtained from KPL, Inc. (Gaithersburg, MD, USA),
which was used at a dilution of 1:8,000. Cell lysis buffer for
western blotting, bicinchoninic acid (BCA) protein assay kit and
SDS-PAGE gel preparation kit were purchased from Beyotime Institute
of Biotechnology (Haimen, China).
Diabetes model
The rats were allowed to acclimatize for 7 days
prior to the experiment. Diabetes was induced by a single
intraperitoneal injection of STZ (40 mg/kg) dissolved in 0.1 M
sodium citrate buffer (pH 4.4; prepared from citric acid and
trisodium citrate; Sinopharm Chemical Reagent Co., Ltd., Shanghai,
China) overnight. In place of sterile water, 5% glucose solution
was administered to STZ-treated rats 24 h following injection in
order to prevent death due to hypoglycemic shock. Following STZ
injection (72 h), blood samples were collected from the tail vein
to measure blood glucose levels using a blood glucose meter
(Sinocare Inc., Changsha, China). Only those rats with blood
glucose levels ≥16.8 mM were considered to be successful models of
diabetes and were recruited into the study (5). The rats with lower glucose levels
were subjected to another injection of STZ (20 mg/kg) until their
blood glucose levels were ≥16.8mM
Experimental protocol
The experimental rats were divided into four groups
as follows (n=10 per group): i) Control group (normal rats); ii)
diabetes model (DM) group (diabetes rats); iii) DM + NaHS group
(diabetes rats treated with NaHS); and iv) NaHS group (normal rats
treated with NaHS). The control and DM groups were
intraperitoneally injected with physiological saline for 8 weeks
and the DM + NaHS and NaHS groups were intraperitoneally
administered with an equivalent volume of NaHS at a dose of 100
µmol/kg for 8 weeks. At the end of the experiment, rats were
sacrificed by anesthesia with an intraperitoneal injection of
chloral hydrate (350 mg/kg; Pharmacy of the Affiliated South China
Hospital of the University of South China, Hengyang, China). Hearts
were lavaged with ice-cold normal saline and removed.
Histopathological examination
Myocardium samples from the experimental rats were
fixed using 4% paraformaldehyde (Sinopharm Chemical Reagent Co.),
dehydrated with alcohol, embedded in paraffin (Sinopharm Chemical
Reagent Co.) and cut into 5-µm sections. The sections were
stained using a hematoxylin and eosin (H&E) staining kit
(Beyotime Institute of Biotechnology) or a Masson staining kit
(Nanjing Senbeijia Biological Technology Co., Ltd., Nanjing,
China), and observed under light microscopy (Motic BA210; Motic
Medical Diagnostic Systems Co., Ltd., Xiamen, China) at a
magnification of ×200.
Hydroxyproline content assay
Myocardial hydroxyproline content was measured by a
basic hydrolysis method with a hydroxyproline detection kit
according to the manufacturer's instructions. The left ventricular
tissue of the rats was cut into 10-mg pieces and added to 1 ml
basic hydrolysates. The samples were hydrolyzed at 100°C for 20
min. Absorbance was measured at a wavelength of 490 nm using an
automatic enzyme mark reading meter (cat. no. 500; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and the hydroxyproline
content was calculated. Results were expressed as micrograms of
hydroxyproline per milligram of cardiac tissue (wet weight).
Transmission electron microscopy
analysis
The left ventricular tissue from the harvested
hearts in each group was cut into small pieces on ice and fixed in
2.5% glutaraldehyde (Sinopharm Chemical Reagent Co.), post-fixed in
1% osmium tetroxide (Absin Bioscience Inc., Shanghai, China) and
dehydrated in a series of graded ethanol solutions. Ultrathin
sections were cut and stained with uranyl acetate (Shanghai Fortune
Biological Technology Co., Ltd., Shanghai, China) and lead citrate
(Tanyun Industry Fine Chemical Co., Ltd., Yingkou, China). Samples
were observed and images were captured by transmission electron
microscopy.
Western blot analysis
Total proteins were extracted in ice-cold
radioimmunoprecipitation assay buffer containing protease
inhibitors (Beyotime Institute of Biotechnology), and quantified
using a BCA protein assay kit. The proteins were denatured,
separated by 10% SDS-PAGE electrophoresis and transferred to a
polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA).
The membranes were blocked with 5% skimmed milk in Tris-buffered
saline (Tris supplied by Biosharp Co., Hefei, China and NaCl from
Sinopharm Chemical Reagent Co.) with Tween 20 (Wellbiology Co.,
Ltd. Changsha, China) (TBST) prior to overnight incubation at 4°C
with antibodies against CSE (1:1,000), collagen I (1:400), collagen
III (1:400), MMP7 (1:400), MMP8 (1:400), MMP14 (1:400), TIMP1
(1:400), TGFβ1 (1:400), PI3K (1:400), AKT1 (1:400), Beclin-1
(1:1,000), Atg3 (1:1,000), Atg5 (1:1,000) and Atg16 (1:1,000).
Following washing three times with TBST, the membranes were
incubated with horseradish peroxidase-conjugated secondary antibody
(1:8,000) for 1 h at room temperature. Bands were visualized using
an enhanced chemiluminescence detection reagent (Beyotime Institute
of Biotechnology) and analyzed with a Molecular Imager VersaDoc MP
5000 system (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical differences among the groups were assessed by one-way
analysis of variance with SPSS software, version 18.0 (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
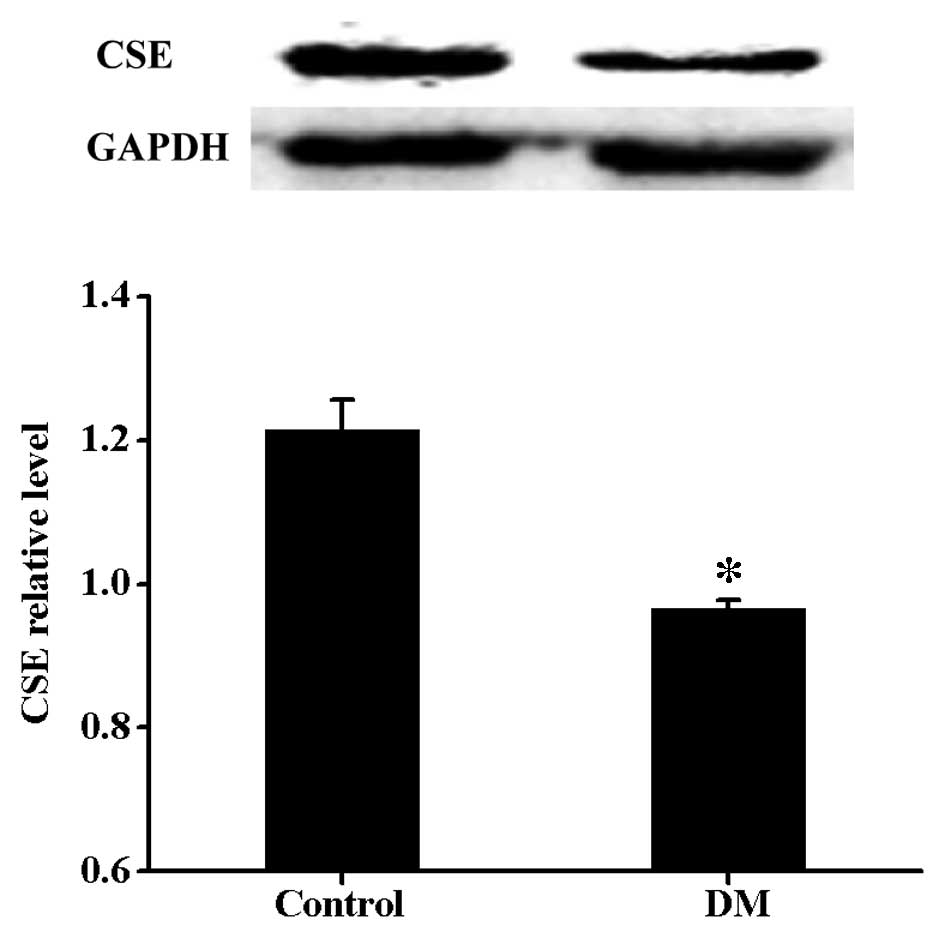
Expression levels of CSE are decreased in
diabetic rat myocardium
To investigate whether diabetes-induced myocardial
damage was associated with decreased generation of endogenous
H2S, the expression level of CSE was measured. As shown
in Fig. 1, compared with the
control group, there was a significant reduction in CSE in the DM
group.
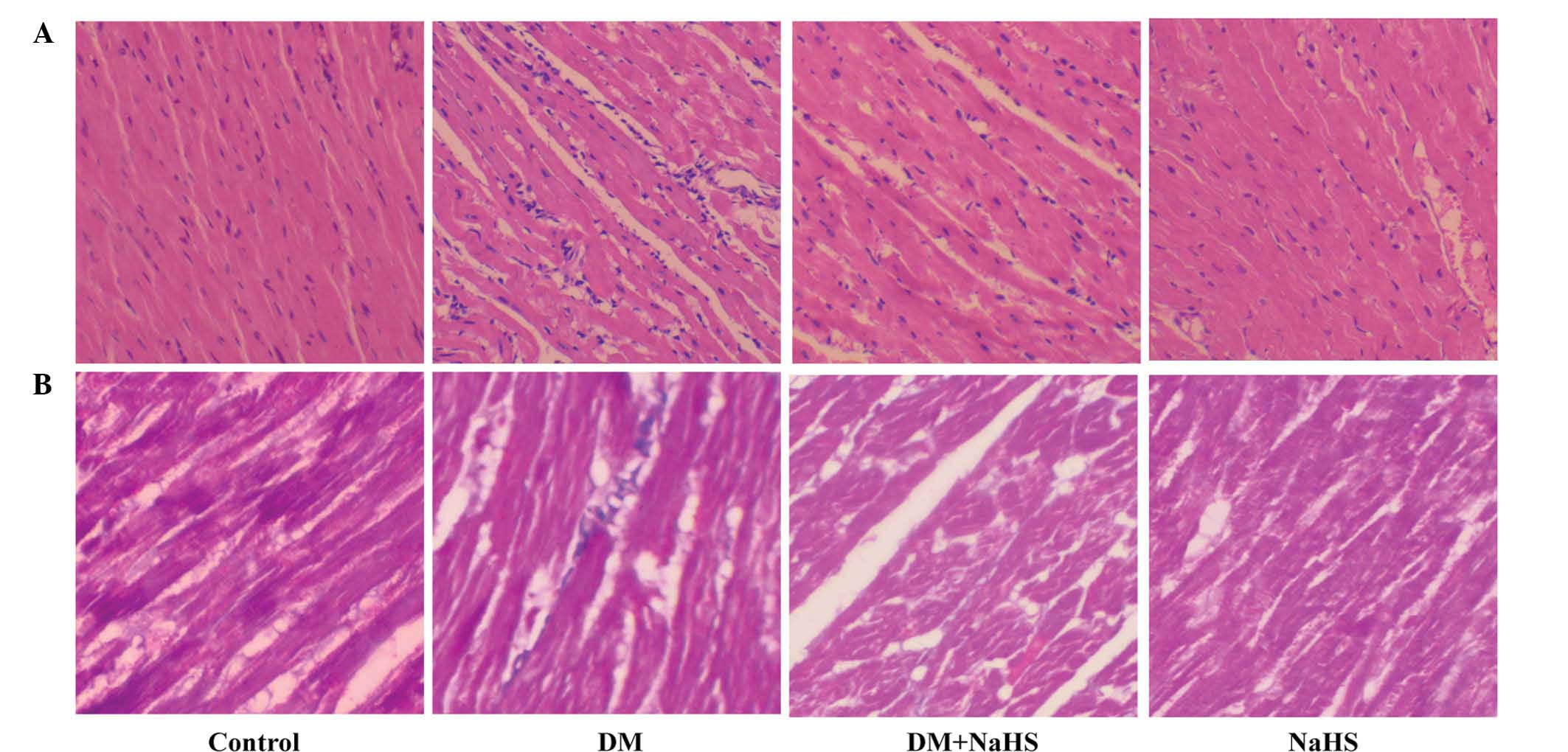
Effect of H2S on
diabetes-induced changes in myocardial morphology
In order to evaluate the morphological changes in
the myocardium, H&E and Masson staining were conducted at the
end of the experiment. Fig. 2
shows the morphological changes observed in the myocardium of the
different groups. The H&E staining (Fig. 2A) demonstrated that in the control
group, the myocardial cells were orderly and compactly arranged,
and less extracellular matrix was observed. In the DM group, the
myocyte cross-sectional area was increased, and relatively
disorganized myocardial cells and increased interstitial ECM were
observed. However, these above-mentioned changes were markedly
reversed in the DM + NaHS group. The results from Masson staining
demonstrated the deposition of collagen fibers (Fig. 2B). Blue staining indicated the
intensity of fibrosis in the cardiac tissue. There was little
evident cardiac fibrosis in the control group, however, increased
fibrosis was observed in DM group. Following NaHS treatment, the
myocardial fibrosis was markedly alleviated compared with the DM
group. H&E and Masson staining demonstrated that, compared with
the control group, there were no notable histological changes in
the NaHS group.
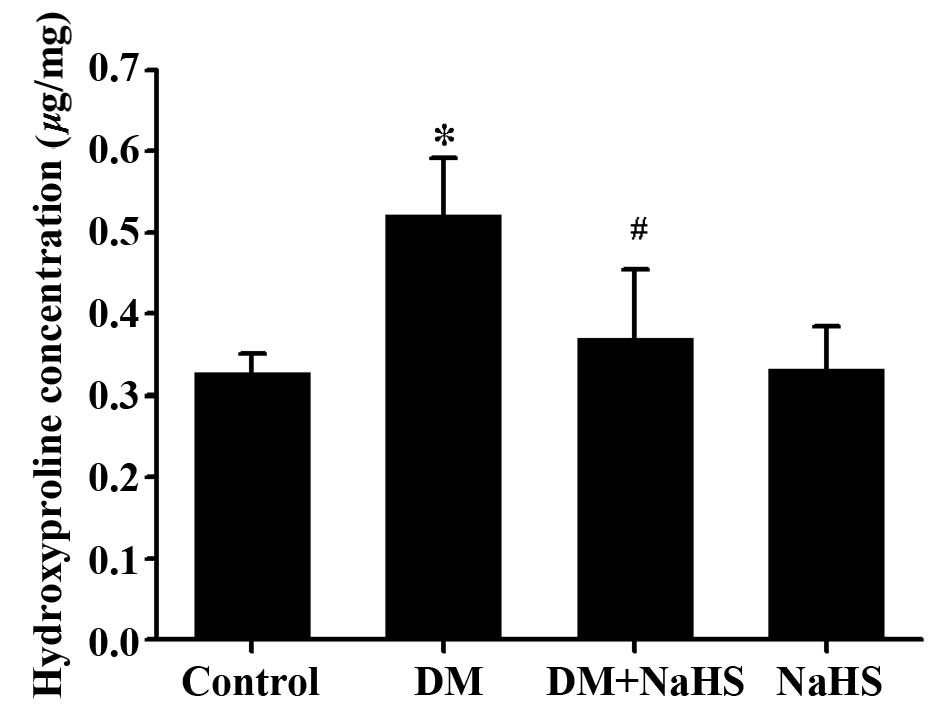
Effect of H2S on
diabetes-induced change in the hydroxyproline content
As a characteristic of collagen production,
hydroxyproline content was determined in the present study
(Fig. 3). The myocardial
hydroxyproline content in the DM group was significantly higher
than that in control group, and it was markedly decreased in the DM
+ NaHS group compared with the DM group. No significant difference
was observed in myocardial hydroxyproline content between the
control group and NaHS group.
Effect of H2S on
diabetes-induced changes in protein expression levels of collagen I
and collagen III
Collagen I and collagen III are the predominant
collagen types in the heart, and the effects of H2S on
the protein expression levels of collagen I and collagen III were
investigated using western blotting (Fig. 4). It was observed that the
expression levels of collagen I and collagen III were increased in
the myocardium of the DM group compared with those in the control
group, while they were reduced in the DM + NaHS group. No
significant differences in the expression of collagen I and
collagen III were examined between control group and the NaHS
group.
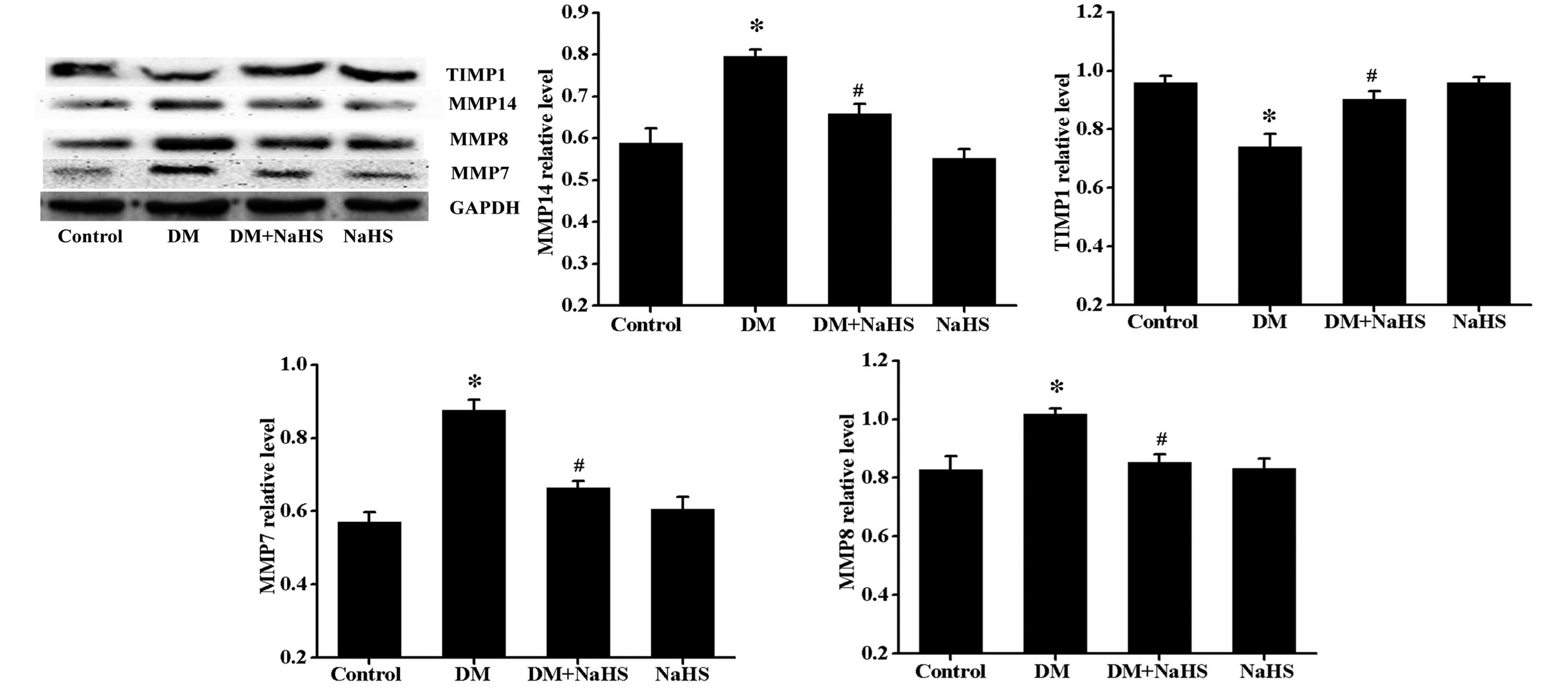
Effect of H2S on
diabetes-induced changes in the expression levels of MMP7, MMP8,
MMP14 and TIMP1
The balance of MMPs and TIMPs determines the ratio
of collagen synthesis and degradation, thus, as it somewhat
indicates the status of fibrosis, the expression levels of MMP7,
MMP8, MMP14 and TIMP1 were determined (Fig. 5). Results of the western blot
analysis indicate that diabetes induced a significant increase in
expression levels of MMP7, MMP8 and MMP14, but a decrease in
expression levels of TIMP1, and that these changes were markedly
reversed by NaHS treatment. No significant difference was observed
in the expression of MMP7, MMP8, MMP14 and TIMP1 between the
control and NaHS groups.
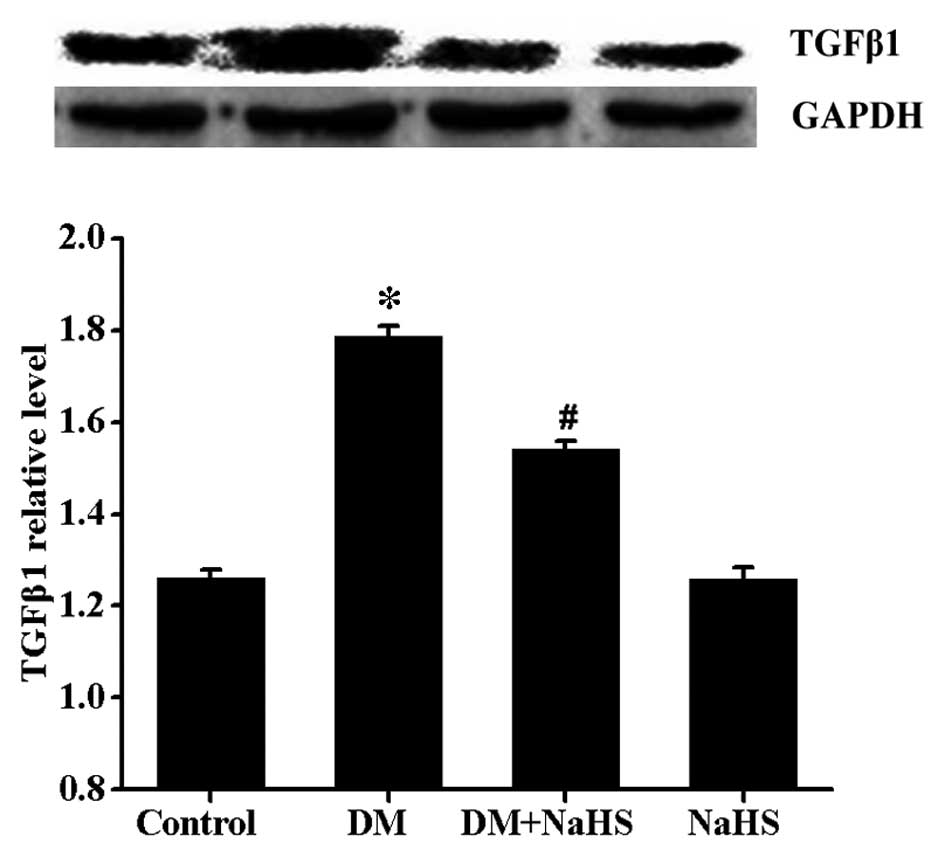
Effect of H2S on
diabetes-induced changes in the expression of TGFβ1
It is widely accepted that TGFβ1 is closely
associated with fibrogenesis. In the present study, the expression
levels of TGFβ1 were determined by western blot analysis (Fig. 6). Results indicated that the
expression levels of TGFβ1 were markedly increased in the DM group
compared with the control group. However, a significant decrease in
TGFβ1 expression was observed in the DM + NaHS group. No
significant difference was observed in the expression of TGFβ1
between the control and NaHS groups.
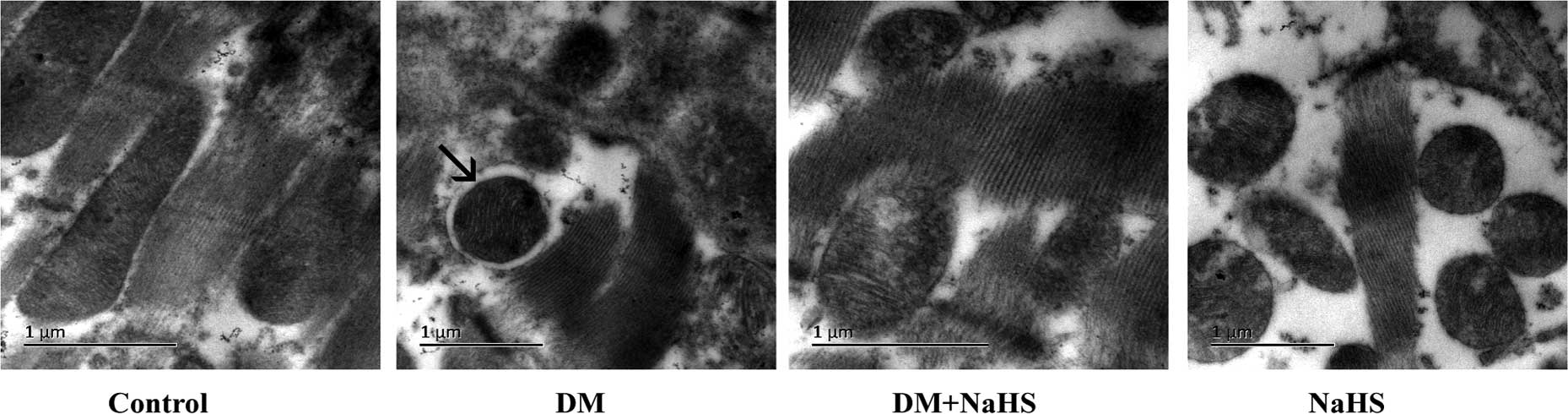
Effect of H2S on
diabetes-induced change in the formation of autophagosomes
To investigate the internal mechanism underlying the
beneficial effects of H2S against cardiomyopathy,
transmission electron microscopy was adopted to observe
autophagosomes (Fig. 7). As
demonstrated in Fig. 7, in the DM
group, autophagosomes were identified in the myocardium, but not in
the DM + NaHS group. In addition, no autophagosomes were observed
in the control or NaHS groups.
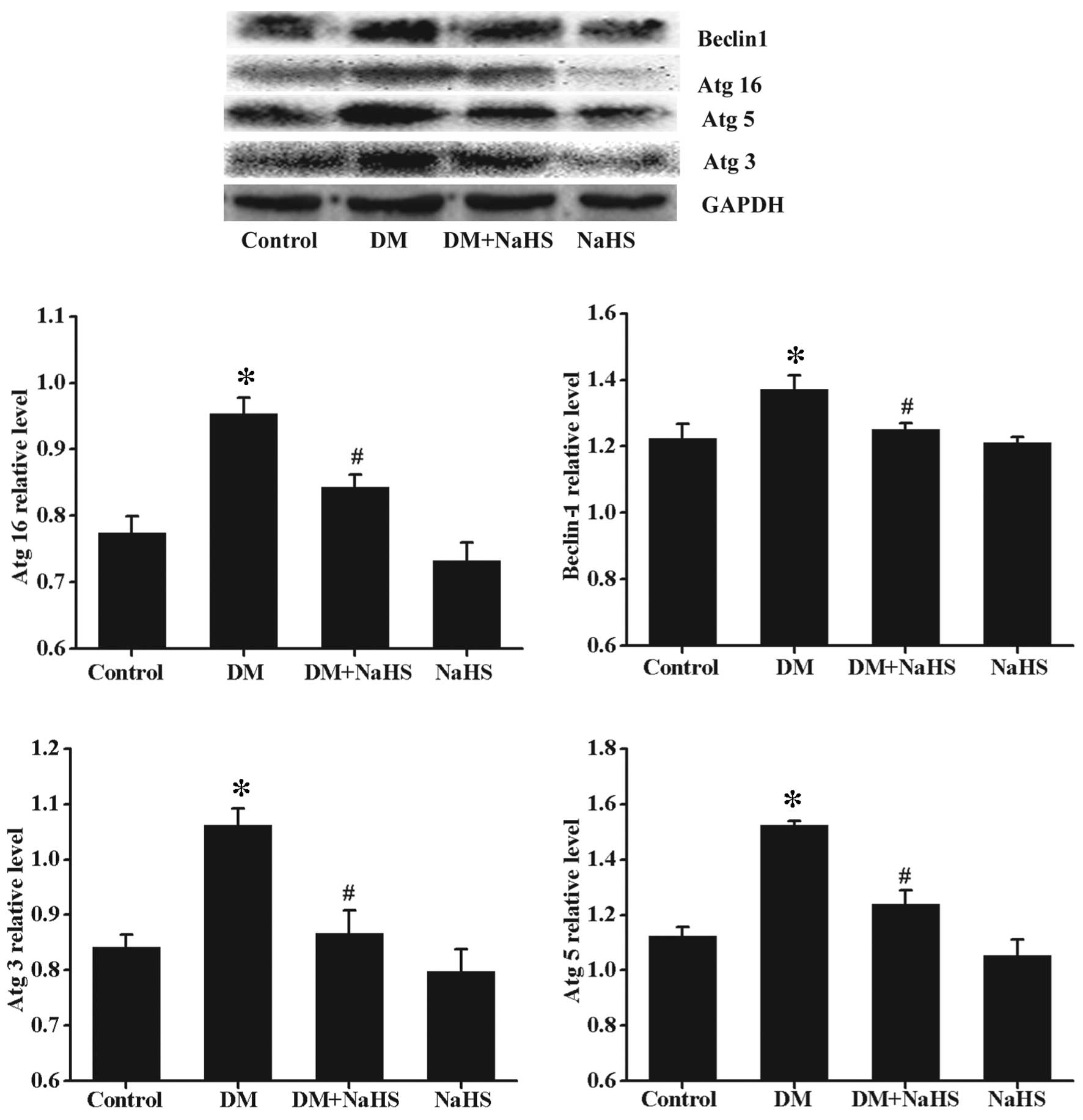
Effect of H2S on
diabetes-induced changes in the expression of autophagy protein
markers
In order to further investigate the above results,
the expression levels of certain autophagy markers were determined
using western blotting (Fig. 8).
Compared with the control group, the expression levels of Beclin-1,
Atg3, Atg5 and Atg16 were significantly upregulated in the DM
group. These autophagy markers were significantly reduced in the DM
+ NaHS group compared with the DM group. No significant difference
was observed in the expression levels of these autophagy markers
between the control and NaHS groups.
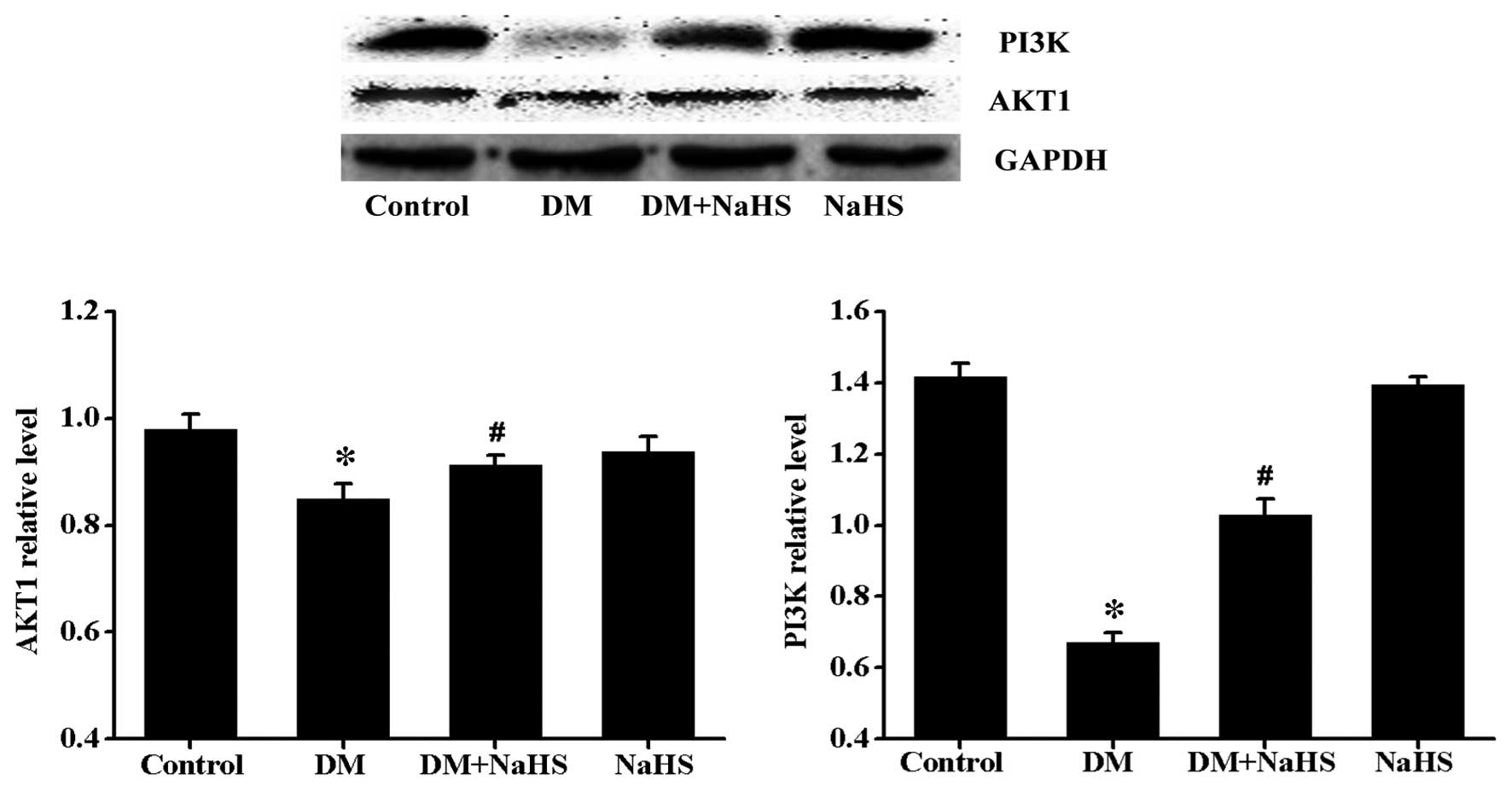
Effects of H2S on
diabetes-induced change in PI3K/AKT1 signaling
To further investigate the potential signaling
pathways involved in diabetes and/or H2S-induced cardiac
autophagic response, the expression levels of PI3K and its
downstream signaling molecule, AKT1 were determined (Fig. 9). Results from the western blot
analysis demonstrated that the expression of PI3K and AKT1 were
significantly lower in the DM group than in the control group,
however, a significant increase in the expression level of PI3K and
AKT1 were observed in the DM + NaHS group. No significant
difference was observed in the expression levels of PI3K and AKT1
between the control and NaHS groups.
Discussion
Prolonged hyperglycemia is hypothesized to induce
metabolic disturbances and result in alterations to the balance of
circulating hormones, which leads to the structural remodeling of
the heart, including fibrosis (21). ECM is important in maintaining left
ventricular geometry and ventricular function (22), and the suppression of ECM
remodeling may be a therapeutic strategy to alleviate the
progression of DCM. In the present study, rat models of diabetes
were induced by intraperitoneal injection of STZ. In diabetic rats,
symptoms of polyuria, polydipsia and polyphagia were observed, as
well as blood glucose levels of ≥16.8 mM, indicating that the
diabetic model was successfully induced. At the end of the
experiment, H&E and Masson staining were performed to evaluate
the degree of fibrosis. The results demonstrated that diabetes
increased myocyte cross-sectional area, induced a certain amount of
disorganization of myocardial cell alignment and enhanced the
deposition of collagen in the DM group. This suggests that
myocardial damage was due to diabetes in the present study.
H2S is an important endogenous signaling
molecule involved in the regulation of the cardiovascular system.
Previous studies have demonstrated the cardioprotective role of
H2S in various models of cardiac injury, including
ischemia/reperfusion injury, isoproterenol-induced heart failure
and left ventricular remodeling induced by chronic alcohol
consumption (23–25). Furthermore, in vitro
research has demonstrated that H2S protects against
high-glucose-induced apoptosis in neonatal rat cardiomyocytes
(26). Based on these findings,
the present study hypothesized that administration of NaHS as
H2S donor would be of benefit in DCM. In addition, the
current study demonstrated that the expression of CSE was
significantly decreased in the DM group. CSE is the key enzyme that
cleaves L-cysteine to release H2S in heart, and
downregulation of CSE results in decreased endogenous
H2S generation (6). It
has been further suggested that NaHS treatment may alleviate
myocardial damage induced by diabetes.
In the present study, previously described
pathological abnormalities of the myocardium, which were induced by
diabetes, were notably reversed following NaHS treatment,
suggesting that H2S ameliorates myocardial damage in
rats with diabetes. To further investigate this result, the
concentration of hydroxyproline and the expression of collagen I
and collagen III were determined using the basic hydrolysis method
and western blot analysis, respectively. Hydroxyproline content in
the myocardium is used as a quantitative estimation of myocardial
collagen production as it is widely understood that hydroxyproline
constitutes 14% of the total amino acids of collagen (27,28).
It was observed that there were markedly higher levels of
hydroxyproline and expression levels of collagen I and collagen III
in the DM group; however, NaHS administration significantly
downregulated the levels of hydroxyproline, collagen I and collagen
III. These results also suggest that diabetes is associated with
marked myocardial fibrosis and that H2S exerts a
protective effect on the normal myocardial structure.
MMPs and their endogenous inhibitors, TIMPs are the
primary coordinators of collagen synthesis and degradation in
cardiac tissue (29). MMPs are a
family of enzymes that cleave matrix components and degrade
fibrillar collagen, however, the products of degraded proteins
serve as stimulators for collagen synthesis (30,31).
Previous studies have demonstrated that increased MMP expression
levels and decreased expression levels of TIMP are accompanied by
increased fibrosis, whereas downregulated MMP is associated with
decreased deposition of collagen fibers (32,33).
TGFβ1 is widely accepted as a critical factor in fibrogenesis. It
has been previously reported that TGFβ1 stimulates fibroblast
proliferation and the production of ECM proteins, including
fibronectin and collagen (34).
Previous studies have also demonstrated that elevated expression
levels of TGFβ1 are implicated in multiple fibrotic diseases, such
as liver cirrhosis, pulmonary fibrosis and sclerosis (35–37).
Consistent with the pathology results and altered expression of
fibrosis markers mentioned above, NaHS treatment significantly
alleviates diabetes-induced increases in the levels of MMPs and
TGFβ1, and decrease of TIMP.
Myocardial fibrosis is commonly accompanied by
excessive cardiomyocyte death (38,39);
an increased rate of cardiomyocyte death is characteristic of
cardiac remodeling (40). Cardiac
tissue consists of cardiomyocytes and extracellular matrix,
excessive loss of myocardial cells increases the extracellular
matrix and results in myocardial cells being replaced with fibrotic
tissue. The accelerated death of cardiomyocytes promotes the
progression of myocardial fibrosis.
Autophagy is the endogenous, tightly regulated
cellular 'housekeeping' process responsible for the degradation of
damaged and dysfunctional cellular organelles and protein
aggregates (41). In a
nutrient-deprived cell, autophagy is a cell-survival mechanism
(42). However, prolonged
activation of the autophagic signaling pathway results in cell
death due to excessive self-digestion and degradation of essential
constituents (43). For example,
autophagy is involved in T cell death following the HIV-1 envelope
protein binding to C-X-C chemokine receptor type 4 (44). In human leukemic cells, cell death
resulting from downregulation of Bcl-2 was associated with
increased autophagy (45).
Furthermore, in the heart, autophagy is an essential form of cell
death, along with apoptosis and necrosis (40). A previous study demonstrated that
autophagy, rather than apoptosis, was the major model of
cardiomyocyte death in the UM-X 7.1 hamster model of human dilated
cardiomyopathy. Administration of granulocyte colony-stimulating
factor treatment improved survival, ventricular function and
remodeling, and reduced myocardial fibrosis; these beneficial
effects were possibly associated with a reduction in autophagy
(46). In the load-induced heart
failure model, increased cardiac autophagy was observed. In
addition, the inhibition of expression of Beclin-1, a protein
required for early autopha-gosome formation, decreased
cardiomyocyte autophagy and reduced pathological remodeling. By
contrast, Beclin-1 overexpression increases autophagic activity and
pathological remodeling (47).
Another study regarding myocardial ischemia-reperfusion injury
reported that although induction of autophagy during the ischemic
phase was protective, further increases in autophagy during the
reperfusion phase may induce cell death and appeared to be
detrimental (48). These findings
suggest that autophagy is closely associated with the occurrence of
myocardial injury and the progression of heart failure. In the
present study, diabetes triggered autophagy in the myocardium as
evidenced by the increased number of autophagosomes and the
increased expression of autophagy-associated proteins, the effects
of which were ameliorated by NaHS treatment.
A central checkpoint that negatively regulates
autophagy is mechanistic target of rapamycin, which is the
downstream target of the PI3K/AKT signaling pathway. Upregulation
of the PI3K/AKT1 signaling pathway has been demonstrated to
suppress autophagy (49). In the
present study, the PI3K/AKT1 signaling pathway was markedly
inhibited in the myocardium of the DM group, whereas NaHS treatment
was observed to activate PI3K/AKT1 in diabetic rats, which
suggested that H2S may protect against diabetes-induced
cardiac autophagy via regulation of the PI3K/AKT1 signaling
pathway.
In conclusion, the results of the current study
demonstrated that H2S ameliorated myocardial fibrosis in
rat diabetes models, and that its mechanism may involve inhibition
of excessive activation of autophagy via regulation of PI3K/AKT1
pathway. The results provide a novel theoretical foundation for the
anti-fibrosis mechanism of H2S and suggests novel
targets for DCM therapeutic strategies, although these findings
require further verification. Further research should investigate
whether 5′ AMP-activated protein kinase, another important
regulator of autophagy, is involved in diabetes and/or the
H2S-induced cardiac autophagy response. In vitro
studies using inhibitors to block autophagy or its regulatory
pathway are required to further the present study.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81202830).
Abbreviations:
|
H2S
|
hydrogen sulfide
|
|
NaHS
|
sodium hydrosulfide
|
|
DCM
|
diabetic cardiomyopathy
|
|
ECM
|
extracellular matrix
|
|
CSE
|
cystathionine-γ-lyase
|
|
STZ
|
streptozotocin
|
|
MMPs
|
matrix metalloproteinases
|
References
|
1
|
Pappachan JM, Varughese GI, Sriraman R and
Arunagirinathan G: Diabetic cardiomyopathy: Pathophysiology,
diagnostic evaluation and management. World J Diabetes. 4:177–189.
2013.PubMed/NCBI
|
|
2
|
Asbun J and Villarreal FJ: The
pathogenesis of myocardial fibrosis in the setting of diabetic
cardiomyopathy. J Am Coll Cardiol. 474:693–700. 2006. View Article : Google Scholar
|
|
3
|
Falcão-Pires I and Leite-Moreira AF:
Diabetic cardiomyopathy: Understanding the molecular and cellular
basis to progress in diagnosis and treatment. Heart Fail Rev.
17:325–344. 2012. View Article : Google Scholar
|
|
4
|
Calvent JW, Coetzee WA and Lefer DJ: Novel
insights into hydrogen sulfide-mediated cytoprotection. Antioxid
Redox Sign. 12:1203–1217. 2010. View Article : Google Scholar
|
|
5
|
Yuan P, Xue H, Zhou L, Qu L, Li C, Wang Z,
Ni J, Yu C, Yao T, Huang Y, et al: Rescue of mesangial cells from
high glucose-induced over-proliferation and extracellular matrix
secretion by hydrogen sulfide. Nephrol Dial Transplant.
26:2119–2126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mani S, Li H, Untereiner A, Wu L, Yang G,
Austin RC, Dickhout JG, Lhoták Š, Meng QH and Wang R: Decreased
endogenous production of hydrogen sulfide accelerates
atherosclerosis. Circulation. 127:2523–2534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
King AL and Lefer DJ: Cytoprotective
actions of hydrogen sulfide in ischaemia-reperfusion injury. Exp
Physiol. 96:840–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Wang Q, Guo W and Zhu YZ: Hydrogen
sulfide attenuates cardiac dysfunction in a rat model of heart
failure: A mechanism through cardiac mitochondrial protection.
Biosci Rep. 31:87–98. 2011. View Article : Google Scholar
|
|
9
|
Zhao W, Zhang J, Lu Y and Wang R: The
vasorelaxant effect of H(2)S as a novel endogenous
gaseous K(ATP) channel opener. EMBO J. 20:6008–6016. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao W, Ndisang JF and Wang R: Modulation
of endogenous production of H2S in rat tissues. Can J
Physiol Pharmacol. 81:848–853. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kang B, Hong J, Xiao J, Zhu X, Ni X, Zhang
Y, He B and Wang Z: Involvement of miR-1 in the protective effect
of hydrogen sulfide against cardiomyocyte apoptosis induced by
ischemia/reperfusion. Mol Biol Rep. 41:6845–6853. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Elrod JW, Calvert JW, Morrison J, Doeller
JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al:
Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury
by preservation of mitochondrial function. Proc Natl Acad Sci USA.
104:15560–15565. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calvert JW, Elston M, Nicholson CK,
Gundewar S, Jha S, Elrod JW, Ramachandran A and Lefer DJ: Genetic
and pharmacologic hydrogen sulfide therapy attenuates
ischemia-induced heart failure in mice. Circulation. 122:11–19.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou X, Feng Y, Zhan ZB and Chen JC:
Hydrogen sulfide alleviates diabetic nephropathy in a
streptozotocin-induced diabetic rat model. J Biol Chem.
289:28827–28834. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou X, An G and Chen J: Hydrogen sulfide
improves left ventricular function in smoking rats via regulation
of apoptosis and autophagy. Apoptosis. 19:998–1005. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levine B and Yuan J: Autophagy in cell
death: An innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Younce CW, Wang K and Kolattukudy PE:
Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1
production and induction of a novel zinc-finger protein MCPIP.
Cardiovasc Res. 87:665–674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhu H, Rothermel BA and Hill JA: Autophagy
in load-induced heart disease. Methods Enzymol. 453:343–363. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang L, Yuan T, Du G, Zhao Q, Ma L and Zhu
J: The impact of 1,25-dihydroxyvitamin D3 on the expression of
connective tissue growth factor and transforming growth factor-β1
in the myocardium of rats with diabetes. Diabetes Res Clin Pract.
104:226–233. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Lv H, Gu Y, Wang X, Cao H, Tang Y,
Chen H and Huang C: Protective effect of lycopene on cardiac
function and myocardial fibrosis after acute myocardial infarction
in rats via the modulation of p38 and MMP-9. J Mol Histol.
45:113–120. 2014. View Article : Google Scholar
|
|
23
|
Andreadou I, Iliodromitis EK, Rassaf T,
Schulz R, Papapetropoulos A and Ferdinandy P: The role of
gasotransmitters NO, H2S and CO in myocardial
ischaemia/reperfusion injury and cardioprotection by
preconditioning, postconditioning and remote conditioning. Br J
Pharmacol. 172:1587–1606. 2014. View Article : Google Scholar
|
|
24
|
Liu YH, Lu M, Xie ZZ, Hua F, Xie L, Gao
JH, Koh YH and Bian JS: Hydrogen sulfide prevents heart failure
development via inhibition of renin release from mast cells in
isoproterenol-treated rats. Antioxid Redox Signal. 20:759–769.
2014. View Article : Google Scholar
|
|
25
|
Zhou X, Lu X, Xu W and Chen J: Protective
effects of hydrogen sulfide against chronic alcohol intake-induced
left ventricular remodeling in rats. Cardiovasc Drugs Ther.
27:221–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou X and Lu X: Hydrogen sulfide inhibits
high-glucose-induced apoptosis in neonatal rat cardiomyocytes. Exp
Biol Med (Maywood). 238:370–374. 2013. View Article : Google Scholar
|
|
27
|
Xu X, Ding F, Pang J, Gao X, Xu RK, Hao W,
Cao JM and Chen C: Chronic administration of hexarelin attenuates
cardiac fibrosis in the spontaneously hypertensive rat. Am J
Physiol Heart Circ Physiol. 303:H703–H711. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bish LT, Yarchoan M, Sleeper MM, Gazzara
JA, Morine KJ, Acosta P, Barton ER and Sweeney HL: Chronic losartan
administration reduces mortality and preserves cardiac but not
skeletal muscle function in dystrophic mice. PLoS One.
6:e208562011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li YY, McTiernan CF and Feldman AM:
Interplay of matrix metalloproteinases, tissue inhibitors of
metalloproteinases and their regulators in cardiac matrix
remodeling. Cardiovasc Res. 46:214–224. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li YY, Feng YQ, Kadokami T, McTiernan CF,
Draviam R, Watkins SC and Feldman AM: Myocardial extracellular
matrix remodeling in transgenic mice overexpressing tumor necrosis
factor alpha can be modulated by anti-tumor necrosis factor alpha
therapy. Proc Natl Acad Sci USA. 97:12746–12751. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maquart FX, Bellon G, Chaqour B, Wegrowski
J, Patt LM, Trachy RE, Monboisse JC, Chastang F, Birembaut P and
Gillery P: In vivo stimulation of connective tissue accumulation by
the tripeptide-copper complex
glycyl-L-histidyl-L-lysine-Cu2+ in rat experimental
wounds. J Clin Invest. 92:2368–2376. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dixon IM, Ju H, Reid NL, Scammell-La Fleur
T, Werner JP and Jasmin G: Cardiac collagen remodeling in the
cardiomyopathic Syrian hamster and the effect of losartan. J Mol
Cell Cardiol. 29:1837–1850. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cowan KN, Jones PL and Rabinovitch M:
Regression of hypertrophied rat pulmonary arteries in organ culture
is associated with suppression of proteolytic activity, inhibition
of tenascin-C, and smooth muscle cell apoptosis. Circ Res.
84:1223–1233. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Spiekman M, Przybyt E, Plantinga JA, Gibbs
S, van der Lei B and Harmsen MC: Adipose tissue-derived stromal
cells inhibit TGF-β1-induced differentiation of human dermal
fibroblasts and keloid scar-derived fibroblasts in a paracrine
fashion. Plast Reconstr Surg. 134:699–712. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee WR, Kim KH, An HJ, Kim JY, Lee SJ, Han
SM, Pak SC and Park KK: Apamin inhibits hepatic fibrosis through
suppression of transforming growth factor β1-induced hepatocyte
epithelial-mesenchymal transition. Biochem Biophys Res Commun.
450:195–201. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harris WT, Kelly DR, Zhou Y, Wang D,
MacEwen M, Hagood JS, Clancy JP, Ambalavanan N and Sorscher EJ:
Myofibroblast differentiation and enhanced TGF-B signaling in
cystic fibrosis lung disease. PLoS One. 8:e701962013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Masola V, Zaza G, Secchi MF, Gambaro G,
Lupo A and Onisto M: Heparanase is a key player in renal fibrosis
by regulating TGF-β expression and activity. Biochim Biophys Acta.
1843:2122–2128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Radovits T, Korkmaz S, Mátyás C, Oláh A,
Németh BT, Páli S, Hirschberg K, Zubarevich A, Gwanmesia PN, Li S,
et al: An altered pattern of myocardial histopathological and
molecular changes underlies the different characteristics of type-1
and type-2 diabetic cardiac dysfunction. J Diabetes Res.
2015:7287412015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yu X, Zhang Q, Cui W, Zeng Z, Yang W,
Zhang C, Zhao H, Gao W, Wang X and Luo D: Low molecular weight
fucoidan alleviates cardiac dysfunction in diabetic Goto-Kakizaki
rats by reducing oxidative stress and cardiomyocyte apoptosis. J
Diabetes Res. 2014:4209292014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nishida K and Otsu K: Cell death in heart
failure. Circ J. 72:A17–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dong Y, Undyala VV, Gottlieb RA, Mentzer
RM Jr and Przyklenk K: Autophagy: Definition, molecular machinery,
and potential role in myocardial ischemia-reperfusion injury. J
Cardiovasc Pharmacol Ther. 15:220–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dziedzic SA and Caplan AB: Autophagy
proteins play cyto-protective and cytocidal roles in leucine
starvation-induced cell death in Saccharomyces cerevisiae.
Autophagy. 8:731–738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nishida K, Kyoi S, Yamaguchi O, Sadoshima
J and Otsu K: The role of autophagy in the heart. Cell Death
Differ. 16:31–38. 2009. View Article : Google Scholar
|
|
44
|
Espert L, Denizot M, Grimaldi M,
Robert-Hebmann V, Gay B, Varbanov M, Codogno P and Biard-Piechaczyk
M: Autophagy is involved in T cell death after binding of HIV-1
envelope proteins to CXCR4. J Clin Invest. 116:2161–2172. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saeki K, Yuo A, Okuma E, Yazaki Y, Susin
SA, Kroemer G and Takaku F: Bcl-2 down-regulation causes autophagy
in a caspase-independent manner in human leukemic HL60 cells. Cell
Death Differ. 7:1263–1269. 2000. View Article : Google Scholar
|
|
46
|
Miyata S, Takemura G, Kawase Y, Li Y,
Okada H, Maruyama R, Ushikoshi H, Esaki M, Kanamori H, Li L, et al:
Autophagic cardiomyocyte death in cardiomyopathic hamsters and its
prevention by granulocyte colony-stimulating factor. Am J Pathol.
168:386–397. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matsui Y, Kyoi S, Takagi H, Hsu CP,
Hariharan N, Ago T, Vatner SF and Sadoshima J: Molecular mechanisms
and physiological significance of autophagy during myocardial
ischemia and reperfusion. Autophagy. 4:409–415. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma H, Guo R, Yu L, Zhang Y and Ren J:
Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial
ischaemia/reperfusion injury: Role of autophagy paradox and toxic
aldehyde. Eur Heart J. 32:1025–1038. 2011. View Article : Google Scholar :
|