Introduction
Neuronal cell death is a normal physiological
process during nervous system development. However, it is also a
pathological process in brain and spinal cord disease and injury
(1). Abnormal neuronal cell death
in the central nervous system (CNS) is also associated with acute
neurological injury, such as cerebral ischemia and trauma, as well
as chronic neurodegenerative disorders, such as Alzheimer's disease
and Parkinson's disease (PD). Excitatory neurotransmitter-induced
neurotoxicity, neurotoxins, and oxidative stress are commonly used
to induce neuronal cell death in in vivo and in vitro
models (2–4).
It has been proposed that apoptosis and necrosis are
involved in neuronal cell death, which is caused by excitatory
neurotransmitter-induced neurotoxicity, neurotoxins and oxidative
stress (5). Apoptosis is a
programmed cell death pathway, which is regulated by an
evolutionarily conserved cellular pathway that involves activation
of the caspase cascade (6).
Programmed necrosis (also called necroptosis in cell cultures) is
an essential process of animal development, but also leads to human
diseases (7). Notably, cell death
following neonatal brain injury is caused by programmed necrosis
(8). This indicates the critical
role of programmed necrosis in neuronal cell death following
injury. The mechanism of programmed necrosis has not been fully
elucidated. Receptor-interacting protein (RIP) kinase 1 and RIP
kinase 3, are hypothesized to be the key signaling molecules in
programmed necrosis, and the two may be regulated by caspases and
ubiquitination (9,10). The mechanisms by which neuronal
cells die in response to excitatory neurotransmitter-induced
neurotoxicity, neurotoxins and oxidative stress have not been
determined.
In the present study, the mechanism by which human
SH-SY5Y neuroblastoma cells die in response to glutamate-induced
neurotoxicity, 6-hydroxydopamine (6-OHDA)-induced neurodegenerative
toxicity and glucose oxidase-induced oxidative stress was
investigated. The findings may provide the basis for understanding
the mechanism by which neuronal cells die in response to these
types of stimuli.
Materials and methods
Reagents
Glutamate, 6-OHDA, glucose oxidase and
dithiothreitol were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine
serum (FBS) were obtained from GE Healthcare Life Sciences (Logan,
UT, USA). BD™ MitoScreen (JC-1) and an Annexin V/propidium iodide
(PI) kit were purchased from BD Biosciences (San Jose, CA, USA). A
calcium fluorescent probe (Fluo-3 AM) was purchased from Beyotime
Institute of Biotechnology (Wuhan, China). Mouse anti-human
anti-RIP kinase 1 (ab56815) and rabbit anti-human anti-RIP kinase 3
(ab180535) primary antibodies were obtained from Abcam (Cambridge,
UK). Alexa Fluor 680-conjugated goat anti-mouse immunoglobulin G
(IgG) (A28183) and Alexa Fluor 790-conjugated goat anti-mouse IgG
(A28182) secondary antibodies were purchased from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA), and tetramethylethylenediamine
was purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
Chemical reagents used for western blot analysis, including
glycine, bisacrylamide, and acrylamide were obtained from Wako Pure
Chemical Industries, Ltd. (Osaka, Japan), and ammonium persulfate
was obtained from Sangon Biotech Co., Ltd.
Cell culture
Human SH-SY5Y neuroblastoma cells were obtained from
the China Center for Type Cultural Collection (Wuhan University,
Wuhan, China). The cells were cultured in DMEM culture medium
supplemented with 10% FBS and maintained at 37°C in a 5%
CO2 humidified atmosphere. Upon reaching 80% confluence,
the cells were digested with 0.25% trypsin for 3 min until the
majority of the cells detached. The cells were subsequently
resuspended with DMEM culture medium containing 10% FBS. Following
centrifuging at 97 × g for 10 min, the supernatant was removed and
cells were resus-pended with fresh culture medium. The cell density
was adjusted to 5×105 cells per well for each 6-well
plate or 1×104 cells per well for each 96-well plate.
Subculturing was performed every three or four days, upon the cells
reaching 80% confluence.
Glutamate neurotoxicity model
One day after initial seeding, cells were exposed to
different concentrations of glutamate (10, 15, 20, 25 or 50 mM)
diluted in serum-free DMEM culture medium. Non-treated cells served
as a negative control. The culture medium was removed following an
exposure time of 2, 4, 6 or 8 h. The cells were washed twice with
phosphate-buffered saline (PBS) or distilled water and collected
for subsequent experiments.
6-OHDA-induced neurodegenerative toxicity
model
One day after initial seeding, cells were exposed to
varying concentrations of 6-OHDA (0.05, 0.25, 0.5, 1 or 2 mM)
dissolved in serum-free DMEM culture medium. Non-treated cells
served as a negative control. The culture medium was removed
following an exposure time of 4, 8 or 12 h. The cells were washed
twice with PBS or distilled water and collected for subsequent
experiments.
Glucose oxidase oxidative stress
model
One day after initial seeding, cells were exposed to
different concentrations of glucose oxidase (0.005, 0.05, 0.5, 5 or
20 mg/ml) dissolved in serum-free DMEM culture medium. Non-treated
cells served as a negative control. The culture medium was removed
following an exposure time of 10, 20, 40 or 60 min. The cells were
washed twice with PBS or distilled water and collected for
subsequent experiments.
Determination of cell viability
Cells were seeded into 96-well plates
(1×104 cells per well). Subsequent to the appropriate
incubation time with the indicated compound concentrations, 100
µl MTT (0.5 mg/ml; Sangon Biotech Co., Ltd.) was added to
each well. The cells were incubated for 4 h at 37°C, the medium was
removed and 150 µl dimethyl sulfoxide (Sangon Biotech Co.,
Ltd.) was added to each well to resuspend the MTT metabolic
product. The absorbance of the dissolved formazan was measured at
490 nm with a microplate reader (Model 550; Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Data were calculated from three
independent experiments.
Evaluation of cell apoptosis and
necrosis
Cells were seeded into 6-well plates
(5×105 cells per well). Following the appropriate
incubation period, the culture medium was removed and cells were
washed with PBS. Cells were collected by centrifuging at 67 × g for
5 min. Following PBS washing, 3×104 cells were
resuspended with the staining solution containing 5 µl PI
and 5 µl Annexin V. The samples were examined by flow
cytometry (BD FACSCalibur™; BD Biosciences) after a 30-min
incubation in the dark. Annexin V-positive/PI-negative cells were
identified as early apoptotic cells, and Annexin
V-positive/PI-positive cells were identified as late apoptotic and
necrotic cells.
Assessment of intracellular calcium ion
concentration
Cells were seeded into 6-well plates
(5×105 cells per well). Subsequent to the appropriate
incubation period, the culture medium was removed and cells were
washed twice with distilled water. Cells were probed with 500
µl Fluo-3a solution for 1 h at 37°C. Following incubation,
the Fluo-3a solution was removed and 2 ml distilled water was added
to resuspend the cells. Cells were collected by centrifuging at 67
× g for 5 min and resuspended with distilled water. The samples
were examined by flow cytometry, using the BD FACSCalibur™.
Analysis of mitochondrial membrane
potential
Cells were seeded into 6-well plates
(5×105 cells per well). Following the appropriate
incubation period, the culture medium was removed and cells were
washed twice with PBS. Cells were probed with 500 µl
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolo-carbocyanine
iodide (JC-1) staining solution for 30 min at 37°C. Following
incubation, the JC-1 solution was removed and 200 µl PBS was
added to resuspend the cells. The samples were then examined by
flow cytometry, using the BD FACSCalibur™.
Western blot analysis
Cells were seeded into 6-well plates
(5×105 cells per well). Following the appropriate
incubation period, the culture medium was removed and cells were
washed twice with PBS. Total protein was extracted from cells using
radioimmunoprecipitation analysis lysis buffer containing 1 mM
phenylmethylsulfonyl fluoride (both Sangon Biotech Co., Ltd.) and
maintained for 30 min on ice. After centrifuging at 13,160 × g for
4 min at 4°C, the supernatant was carefully collected and stored at
−20°C until use. Protein concentrations (1 µg/µl)
were measured using the BCA Protein Assay kit (Beyotime Institute
of Biotechnology). Equal quantities of protein extracts (50 ng)
were then separated by 12% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred to polyvinylidene fluoride
membranes (Sangon Biotech Co., Ltd.). The membranes were blocked
with Tris-buffered saline plus Tween-20 (TBS-T; 0.1% Tween-20)
containing 5% w/v non-fat dry milk (Sangon Biotech Co., Ltd.), then
incubated with the primary antibodies (dilution, 1:1,000) at room
temperature for 2 h. After washing with TBS-T, the membranes were
incubated with secondary antibodies (dilution, 1:1,000) for 1 or 2
h at room temperature. To quantify the protein level, the
expression bands of the target proteins were analyzed using the
Odyssey Infrared Imaging System (LI-COR Biotechnology, Lincoln, NE,
USA). Densitometric values obtained from western blotting were used
to conduct statistical analysis and the housekeeping protein, GAPDH
served as an internal control.
Statistical analysis
Data were analyzed by SPSS statistical software
package, version 5 (SPSS, Inc., Chicago, IL, USA) and all data are
presented as the mean ± standard deviation. Statistical
significance was determined using the two-way analysis of variance
comparison test and P<0.05 was considered to indicate a
statistically significant difference.
Results
Cell viability is decreased by high doses
of glutamate, 6-OHDA and glucose oxidase
The effect of glutamate, 6-OHDA and glucose oxidase
on cell viability was examined. A significant reduction of cell
viability was detected when cells were exposed to glutamate doses
>20 mM (Fig. 1A; P<0.05).
Cell viability was significantly decreased in cells exposed to
concentrations of 6-OHDA >0.5 mM at all time points (Fig. 1B; P<0.05). Furthermore, cell
viability was significantly reduced following exposure to >5
mg/ml glucose oxidase (Fig. 1C;
P<0.05). Overall, a prolonged incubation duration or increased
concentration decreased cell viability for the three stimuli that
were assessed in the SH-SY5Y cells.
 | Figure 1Decreased viability of SH-SY5Y cells
following exposure to various stimuli. Cell viability was
determined by the MTT assay. (A) Cells were treated with 0, 10, 15,
20 or 25 mM glutamate for 2, 4, 6 or 8 h. (B) Cells were treated
with 0, 0.05, 0.25, 0.5, 1 or 2 mM 6-OHDA for 4, 8 or 12 h. (C)
Cells were treated with 0, 0.005, 0.05, 0.5, 5 or 20 mg/ml glucose
oxidase for 10, 20, 40 or 60 min. Data are presented as the mean ±
standard deviation of three independent experiments.
*P<0.05 vs. the control (0). OD, optical density;
6-OHDA, 6-hydroxydopamine. |
Differential induction of apoptosis and
necrosis by glutamate, 6-OHDA and glucose oxidase
Apoptosis and necrosis induced by glutamate, 6-OHDA
and glucose oxidase was investigated using Annexin V/PI staining
followed by flow cytometry. Glutamate treatment induced early
apoptosis and necrosis in the SH-SY5Y cells (Fig. 2A). Apoptosis was significantly
induced after cells were exposed to 25 mM glutamate for 1 h
(P<0.05). In addition, exposure to 20 and 50 mM glutamate
significantly increased the percentage of necrotic cells
(P<0.05). The percentage of necrotic cells was significantly
increased when compared with untreated cells after 1 h of
incubation with 1, 2 or 5 mM 6-OHDA (Fig. 2B; P<0.05). By contrast, early
apoptosis was significantly induced following glucose oxidase
incubation (Fig. 2C; P<0.05).
These data indicate that glutamate induced apoptosis and necrosis,
whereas 6-OHDA only induced necrosis and glucose oxidase only
induced early apoptosis.
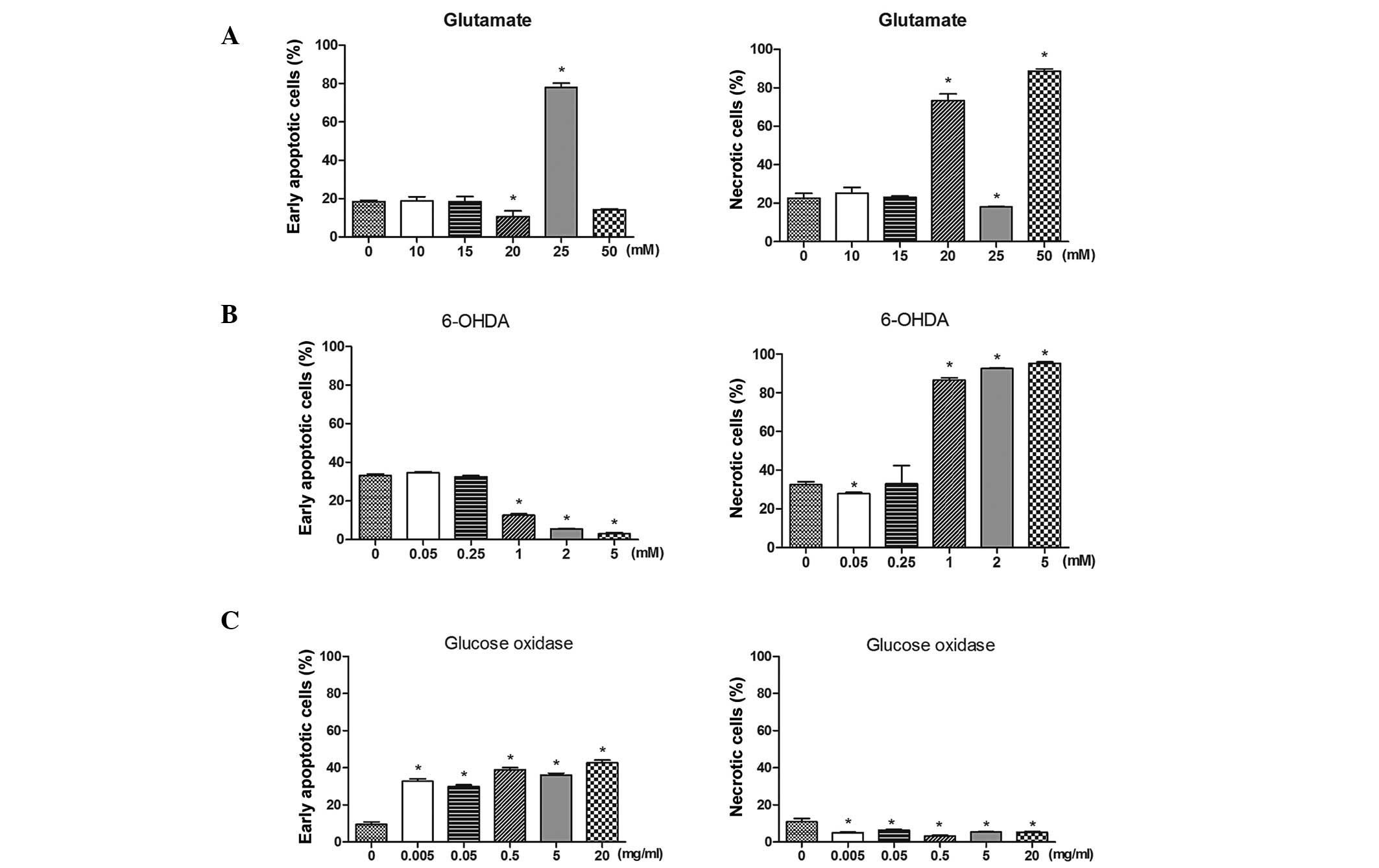 | Figure 2Differential induction of apoptosis
and necrosis of SH-SY5Y cells induced by various stimuli. Cells
were double stained with Annexin V and propidium iodide. The
percentages of early apoptotic or necrotic cells were determined by
flow cytometric analysis. (A) Cells were treated with 0, 10, 15,
20, 25 or 50 mM glutamate for 1 h. (B) Cells were treated with 0,
0.05, 0.25, 1, 2 or 5 mM 6-OHDA for 1 h. (C) Cells were treated
with 0, 0.005, 0.05, 0.5, 5 or 20 mg/ml glucose oxidase for 20 min.
Data are presented as the mean ± standard deviation of three
independent experiments. *P<0.05 vs. the control (0).
6-OHDA, 6-hydroxydopamine. |
Intracellular calcium ion concentration
is increased following exposure to glutamate, 6-OHDA and glucose
oxidase
Cells were stained with Fluo-3a following exposure
to glutamate, 6-OHDA or glucose oxidase to determine the resulting
intracellular calcium ion level. The intracellular calcium
concentration was observed to be dose-dependently elevated
following exposure to glutamate (15–25 mM) for 1 h, (Fig. 3A; P<0.05 vs. the control).
However, no significant difference was identified in the
intracellular calcium concentration of cells treated with 10 or 50
mM glutamate when compared with the control cells (P>0.05).
Furthermore, 6-OHDA (>0.05 mM) and glucose oxidase (>0.05
mg/ml) dose-dependently increased the intracellular calcium
concentration (Fig. 3B and C;
P<0.05). These results indicate that glutamate, 6-OHDA and
glucose oxidase exposure increases intracellular calcium ion
concentrations. Increased intracellular calcium ion concentrations
may result in cell damage.
 | Figure 3Intracellular calcium ion
concentration was increased following exposure to various stimuli.
Following each treatment, cells were stained with Fluo-3a and
analyzed by flow cytometry. The percentages of Fluo-3a-positive
cells are presented. (A) Cells were treated with 0, 10, 15, 20, 25
or 50 mM glutamate for 1 h. (B) Cells were treated with 0, 0.05,
0.25, 0.5, 1 or 2 mM 6-OHDA for 1 h. (C) Cells were treated with 0,
0.005, 0.05, 0.5, 5 or 20 mg/ml glucose oxidase for 20 min. Data
are presented as the mean ± standard deviation of three independent
experiments. *P<0.05 vs. the control (0). 6-OHDA,
6-hydroxydopamine. |
Mitochondrial membrane potential
increased following glutamate, 6-OHDA and glucose oxidase
exposure
The changes in mitochondrial membrane potential of
cells exposed to glutamate, 6-OHDA and glucose oxidase were
determined by JC-1 staining. As shown in Fig. 4A, low doses of glutamate (10 or 15
mM) significantly decreased the mitochondrial membrane potential in
SH-SY5Y cells (P<0.05), whereas high doses of glutamate (25 or
50 mM) significantly increased the mitochondrial membrane potential
in the cells (P<0.05). Concentrations of 6-OHDA that were
>0.25 mM and glucose oxidase concentrations that were >0.005
mg/ml significantly increased the mitochondrial membrane potential
of the cells (Fig. 4B and C;
P<0.05). These findings indicate that the mitochondrial membrane
potential response varied depending on the concentration of
glutamate (high or low), whereas 6-OHDA and glucose oxidase
exposure of any concentration increased the mitochondrial membrane
potential in the SH-SY5Y cells. Elevated mitochondrial membrane
potential may contribute to neuronal cell death.
 | Figure 4Mitochondrial membrane potential
following exposure to various stimuli. Following each treatment,
cells were stained with JC-1 and analyzed by flow cytometry. The
percentage of JC-1-positive cells are presented. (A) Cells were
treated with 0, 10, 15, 20, 25 or 50 mM glutamate for 1 h. (B)
Cells were treated with 0, 0.05, 0.25, 0.5, 1 or 2 mM 6-OHDA for 1
h. (C) Cells were treated with 0, 0.005, 0.05, 0.5, 5 or 20 mg/ml
glucose oxidase for 20 min. Data are presented as the mean ±
standard deviation of three independent experiments.
*P<0.05 vs. the control (0). 6-OHDA,
6-hydroxydopamine. |
RIP kinase 1 expression is induced
following glutamate treatment
The expression levels of RIP kinase 1 and RIP kinase
3 (key signaling molecules in necrosis) following exposure to
glutamate, 6-OHDA and glucose oxidase were determined by western
blotting. The expression of RIP kinase 1 was significantly
upregulated in a dose-dependent manner as a result of glutamate
treatment (Fig. 5A; P<0.05).
However, no significant difference was detected in the expression
level of RIP kinase 3 in cells that were treated with glutamate. In
addition, 6-OHDA and glucose oxidase treatment did not alter the
levels of either RIP kinase 1 or 3 (data not shown). This data
indicates that the RIP kinase 1 signaling pathway may be involved
in glutamate-induced excitatory toxicity.
Discussion
Neuronal cells may exhibit varying responses
depending on the type of stimuli. Stimuli, such as excitatory
neurotransmitter-induced neurotoxicity, neurotoxins and oxidative
stress may induce neuronal cell death (2–4). In
the present study, glutamate, 6-OHDA and glucose oxidase were used
to mimic excitatory neurotransmitter-induced neurotoxicity,
neurotoxins, and oxidative stress in human SH-SY5Y neuroblastoma
cells in order to elucidate the intracellular mechanisms that cause
neuronal cell death.
Glutamate is the primary excitatory neurotransmitter
in the brain. It is associated with various CNS disorders, such as
stroke (11), epilepsy (12) and neurodegenerative diseases
(3). In the current study,
glutamate suppressed cell viability at a concentration of 20 mM.
Decreased cell viability was associated with increased
intracellular calcium concentration and enhanced apoptosis, as well
as necrosis. Consistent with the findings of the present study, a
recent study demonstrated that treatment with 20 mM glutamate
induced apoptosis and elevated reactive oxygen species (ROS)
generation in SH-SY5Y cells (13).
RIP kinase 1 and RIP kinase 3 are key signaling molecules in
programmed necrosis (9,10). The current study demonstrates that
glutamate concentrations >20 mM significantly upregulate the
expression of RIP kinase 1, but not RIP kinase 3, indicating that
RIP kinase 1-mediated programmed necrosis may contribute to
neuronal cell death in glutamate-mediated excitatory toxicity. To
the best of our knowledge, there are only a small number of reports
that indicate the involvement of RIP kinase 1 or RIP kinase 3 in
neuronal damage; thus, future studies are required to investigate
the precise mechanism of RIP-regulated neuronal death.
Neuroinflammatory processes are known to be involved
in the pathogenesis of PD (14).
The pro-inflammatory cytokine, 6-OHDA, a hydroxylated analogue of
dopamine that activates an increase in ROS, is widely administered
to establish cell and animal models of PD (15,16).
In the current study, 6-OHDA suppressed cell viability, induced
cell necrosis, elevated intracellular calcium concentrations and
increased the mitochondrial membrane potential in SH-SY5Y cells.
Notably, 6-OHDA treatment markedly elevated the intracellular
calcium concentration compared with glutamate or glucose oxidase
treatment, indicating that calcium influx and the subsequent
intracellular calcium overload may contribute to 6-OHDA-induced
neuronal cell death. Unlike glutamate treatment, the addition of
6-OHDA did not markedly alter the protein expression of RIP kinase
1 and RIP kinase 3 (data not shown), indicating RIPs may not be
involved in 6-OHDA-mediated neuronal cell death. Elevated
intracellular calcium concentration and mitochondrial membrane
potential in SH-SY5Y cells may contribute to necrotic cell death. A
previous study demonstrated that 6-OHDA treatment induced
mitochondrial dysfunction in SH-SY5Y cells by activating the
mitochondrial translocation of c-Jun N-terminal kinase (JNK)
(17). Hence, other molecular
signals, such as those of JNK, may be involved in 6-OHDA-mediated
cytotoxicity in SH-SY5Y cells.
Oxidative stress is a contributing factor in
neurological and neurodegenerative diseases, and is associated with
aging (18). Glucose oxidase
produces ROS and contributes to mitochondrial DNA damage through
the generation of hydrogen peroxide. Glucose oxidase is, therefore,
widely used to induce cellular toxicity by ROS in vitro
(19). However, to the best of our
knowledge, the effects of glucose oxidase on SH-SY5Y cells remain
to be elucidated. In the present study, glucose oxidase treatment
suppressed cell viability, induced early apoptosis, elevated
intracellular calcium concentration and increased the mitochondrial
membrane potential in SH-SY5Y cells. Oxidative stress did not alter
expression of RIP kinase 1 and RIP kinase 3, indicating that these
proteins may not be involved in oxidative stress-induced neuronal
cell death. It is hypothesized that glucose oxidase leads to early
apoptosis, and that increased intracellular calcium levels may
induce irreversible rupture and cell death.
In conclusion, the present study demonstrates the
different responses of SH-SY5Y cells to various stimuli. Thus,
these SH-SY5Y neuronal cells may be utilized for the determination
of therapeutic agent protection in different experimental
paradigms. In addition, RIP kinase 1 may regulate programmed
necrosis in glutamate-mediated excitatory toxicity, although not in
cell damage induced by either 6-OHDA or glucose oxidase. This
indicates that RIP kinase 1 may serve as a therapeutic target in
the treatment of excitatory neurotransmitter-induced neurotoxicity
in disorders of the CNS.
References
|
1
|
Martin LJ: Neuronal cell death in nervous
system development, disease and injury (Review). Int J Mol Med.
7:455–478. 2001.PubMed/NCBI
|
|
2
|
Melo A, Monteiro L, Lima RM, Oliveira DM,
Cerqueira MD and El-Bacha RS: Oxidative stress in neurodegenerative
diseases: Mechanisms and therapeutic perspectives. Oxid Med Cell
Longev. 2011:4671802011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehta A, Prabhakar M, Kumar P, Deshmukh R
and Sharma PL: Excitotoxicity: Bridge to various triggers in
neurodegenerative disorders. Eur J Pharmacol. 698:6–18. 2013.
View Article : Google Scholar
|
|
4
|
Lopes FM, Londero GF, de Medeiros LM, da
Motta LL, Behr GA, de Oliveira VA, Ibrahim M, Moreira JC, de
Oliveira Porciúncula L, da Rocha JB and Klamt F: Evaluation of the
neurotoxic/neuroprotective role of organoselenides using
differentiated human neuroblastoma SH-SY5Y cell line challenged
with 6-hydroxydopamine. Neurotox Res. 22:138–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan J, Lipinski M and Degterev A:
Diversity in the mechanisms of neuronal cell death. Neuron.
40:401–413. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Galluzzi L, Vanden Berghe T,
Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F
and Kroemer G: Programmed necrosis from molecules to health and
disease. Int Rev Cell Mol Biol. 289:1–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chavez-Valdez R, Martin LJ and Northington
FJ: Programmed necrosis: A prominent mechanism of cell death
following neonatal brain injury. Neurol Res Int. 2012:2575632012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vandenabeele P, Declercq W, Van Herreweghe
F and Vanden Berghe T: The role of the kinases RIP1 and RIP3 in
TNF-induced necrosis. Sci Signal. 3:re42010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chan FK and Baehrecke EH: RIP3 finds
partners in crime. Cell. 148:17–18. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hazell AS: Excitotoxic mechanisms in
stroke: An update of concepts and treatment strategies. Neurochem
Int. 50:941–953. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Coulter DA and Eid T: Astrocytic
regulation of glutamate homeostasis in epilepsy. Glia.
60:1215–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nampoothiri M, Reddy ND, John J, Kumar N,
Kutty Nampurath G and Rao Chamallamudi M: Insulin blocks
glutamate-induced neurotoxicity in differentiated SH-SY5Y neuronal
cells. Behav Neurol. 2014:6741642014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tansey MG and Goldberg MS:
Neuroinflammation in Parkinson's disease: Its role in neuronal
death and implications for therapeutic intervention. Neurobiol Dis.
37:510–518. 2010. View Article : Google Scholar :
|
|
15
|
Kopalli SR, Noh SJ, Koppula S and Suh YH:
Methylparaben protects 6-hydroxydopamine-induced neurotoxicity in
SH-SY5Y cells and improved behavioral impairments in mouse model of
Parkinson's disease. Neurotoxicology. 34:25–32. 2013. View Article : Google Scholar
|
|
16
|
Ribeiro RP, Moreira EL, Santos DB, Colle
D, Dos Santos AA, Peres KC, Figueiredo CP and Farina M: Probucol
affords neuroprotection in a 6-OHDA mouse model of Parkinson's
disease. Neurochem Res. 38:660–668. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chambers JW, Howard S and LoGrasso PV:
Blocking c-Jun N-terminal kinase (JNK) translocation to the
mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro
and in vivo. J Biol Chem. 288:1079–1087. 2013. View Article : Google Scholar :
|
|
18
|
Hybertson BM, Gao B, Bose SK and McCord
JM: Oxidative stress in health and disease: The therapeutic
potential of Nrf2 activation. Mol Aspects Med. 32:234–246. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Salazar JJ and Van Houten B: Preferential
mitochondrial DNA injury caused by glucose oxidase as a steady
generator of hydrogen peroxide in human fibroblasts. Mutat Res.
385:139–149. 1997. View Article : Google Scholar
|















