Introduction
Hearing loss is one of the most common birth
defects. According to the World Health Organization, 360,000,000
individuals worldwide have disabling hearing loss (www.who.int/), including 328,000,000 adults and
32,000,000 children. Deafness is a multi-factorial disease,
predominantly induced by genetic or environmental causes or their
interactions (1). It is suggested
that ~50% of deafness is hereditary, which includes syndromic
hearing loss and nonsyndromic hearing loss (NSHL), which comprises
~70% of cases of hereditary deafness (2). Of NSHL, almost 80% of cases is due to
autosomal recessive inheritance, and it has been demonstrated that
autosomal recessive NSHL is associated with >50 genes (3) (http://hereditaryhearingloss.org/), including the gap
junction protein β 2 (GJB2), solute carrier family 26, member 4
(SLC26A4) and mitochondrial DNA 12S rRNA genes. In NSHL, ~50% of
deafness is caused by mutation of the GJB2 gene (4,5), and
in the GJB2 gene, >90 mutations have been identified. GJB2
35delC is the most common mutation in the European population
(6), while 235delC is common in
the Asian population (7). Mutation
of the SLC26A4 gene is the second leading cause of NSHL, which
contains 21 exons and has been identified to contain ~200
mutations, with IVS7-2A>G and 2168A>G being the two most
common mutations in Asian population (8–10).
The 1,555A>G mutation in the mitochondrial DNA 12S rRNA gene was
the first identified genetic cause for aminoglycoside-induced NSHL
(11). Previous molecular
etiological studies have shown that GJB2, SLC26A4 and 12S rRNA are
three common deafness-associated genes in NSHL among the Chinese
population (12–15). Therefore, mutational screening in
these three genes may be effective and beneficial for diagnosing
deafness in this population.
It is common for deaf couples to have children
together; certain couples may have the same disease-causing genetic
mutation. Thus, confirmation of the pathogenic genotype of deaf
patients may assist in determining the genotype of their children.
In families with children affected by NSHL, it is important to
confirm the genotypes of the couples and their deaf children, in
order to determine the risk in their next pregnancy. In the present
study, the GJB2 gene, SLC26A4 gene and 12S rRNA were examined by
Sanger sequencing in 117 deaf patients, including 39 deaf couples
and 39 deaf individuals. Furthermore, seven pregnant women were
offered prenatal diagnosis via amniocentesis at 18–21 weeks
gestation. The aim of the present study was to analyze the genetics
of deafness and provide appropriate genetic counseling about
hearing loss to our patients.
Materials and methods
Subjects
In the present study 117 deaf patients, aged between
3 months and 35 years were recruited. These patients were diagnosed
with nonsyndromic neurosensory deafness at Nanjing Maternity and
Child Health Care Hospital (Nanjing, China) between 2009 and 2013,
with hearing loss of >70 dB. Any patients with systemic disease,
dysgnosia, syndromic deafness or a history of meningitis, otitis
media or trauma of the ear were excluded from the investigations.
The present study was performed in accordance with the declaration
of Helsinki, and was performed following approval from the ethics
committee of Nanjing Maternity and Child Health Care Hospital
(Nanjing Medical University, Nanjing, China). Written informed
consent was obtained from the patient.
DNA extraction
To obtain DNA, 2 ml peripheral blood was collected
and then placed into tubes with ethylene diamine tetraacetic acid
(EDTA; Greiner Bio-One International GmbH). For the pregnant women
offered prenatal diagnosis, 10 ml amniotic fluid was collected at
18–21 weeks gestation, and DNA was extracted using a DNA extraction
kit (Xiamen Zhishan Biological Company, Xiamen, China), according
to manufacturer's protocol, followed by storage at −20°C.
Primer design
Primers were designed, according to those previously
described (13–15), which were used to perform
polymerase chain reaction (PCR) amplification and sequencing. The
primer sequences are listed in Table
I, and were synthesized by Invitrogen (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The coding sequences of GJB2
were amplified, following which GJB2 genetic mutations were
detected by Sanger sequencing (13,16,17).
Furthermore, mitochondrial DNA from 1,428 to 1,890 were also
amplified and 1,555A>G mutation of the mitochondrial 12S rRNA
gene were detected (15,18,19).
The hot-spot regions of the SLC26A4 gene, including exons 5, 7, 8,
10, 17 and 19, were also amplified, of which exons 7 and 8 were
amplified in one amplicon. Therefore, five fragments were amplified
to cover the six exons and flanking sequence, following which the
SLC26A4 gene mutation was detected. For patients with one allelic
variant in the hot-spot regions, the other exons were sequenced one
by one, until two mutant alleles were identified, as previously
described (14,20,21).
 | Table IPCR primers for the GJB2 gene, SLC26A4
gene and mitochondrial DNA 12S rRNA. |
Table I
PCR primers for the GJB2 gene, SLC26A4
gene and mitochondrial DNA 12S rRNA.
| Gene | Exon | PCR primer
(5′–3′) | Product length
(bp) |
|---|
| GJB2 | 2 |
F-TTGGTGTTTGCTCAGGAAGA | 872 |
| |
R-GGTTGCCTCATCCCTCTCAT | |
| SLC26A4 | 5 |
F-CCTATGCAGACACATTGAACATTTG | 442 |
| |
R-TGAGCCTTAATAAGTGGGGTCTTG | |
| 7+8 |
F-CATGGTTTTTCATGTGGGAAGATTC | 502 |
| |
R-AGACTGACTTACTGACTTAATGT | |
| 10 |
F-AAATACTCAGCGAAGGTCTTGC | 250 |
| |
R-CGAGCCTTCCTCTGTTGC | |
| 17 |
F-CCAAGGAACAGTGTGTAGGTC | 423 |
| |
R-CCCATGTATTTGCCCTGTTGC | |
| 19 |
F-TCACTTGAACTTGGGACGCGGA | 383 |
| |
R-CAACAGCTAGACTAGACTTGTG | |
| Mitochondrial 1
allelic | 1,555 |
F-GCAGTAAACTAAGAGTAGAGT | 463 |
| DNA 12S rRNA | |
R-GGCTCTCCTTGCAAAGTTAT | |
PCR amplification
The GoTaq® PCR system was purchased from
Promega Corporation (Madison, WI, USA). The total volume of the PCR
reaction mixture was 50 µl, including 10 µl of 5X
Green GoTaq® Reaction Buffer, 2 µl of 10 mmol/l
dNTP, 1 µl of each (10 µmol/l) primer, 0.25 µl
of 5 U/µl GoTaq® DNA polymerase, 5 µl of
the 20 ng/µl DNA template and 30.75 µl distilled
water. The amplification reaction conditions for exons 7 and 8 of
the SLC26A4 gene were as follows: 95°C for 5 min; followed by 35
cycles of 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec, and
a final step of 72°C for 7 min. For the remaining six exons, the
reaction conditions were as follows: 95°C for 5 min; followed by 35
cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 1 min, and
a final step of 72°C for 7 min. The amplified products were stored
at 4°C.
Sequence analysis
Following PCR, agarose gel electrophoresis was
performed, and the products were purified according to TIANgel Midi
DNA Purification kits (Tiangen Biotech Co., Ltd., Beijing, China),
following which the purified products were sequenced. Sequencing
kits were obtained from Life Technologies (Grand Island, NY, USA).
The reaction system contained 1 µl Bigdye, 3 µl
buffer, 2 µl primers and 5 µl purified products in a
total volume of 20 µl. The reaction conditions were as
follows: 95°C for 1 min; followed by 35 cycles of 95°C for 10 sec,
50°C for 5 sec and 60°C for 4 min. The amplified products were
stored at 4°C. The sequences of the primers are listed in Table I. In addition, the sequencing
products were purified with 100 mM EDTA solution and alcohol
solution. Following purification, the products were dried and were
dissolved in 100% HiDi Formamide (Gibco; Thermo Fisher Scientific,
Inc.). Subsequently, all products were sequenced (forward and
reverse) using an ABI3130 analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The sequence results were aligned with
standard sequences using SeqBuilder procedures on Lasergene
software, version 7.1 (DNASTAR, Inc., Madison, WI, USA). The GJB2,
SLC26A4 and mitochondrial DNA 12S rRNA genetic standards were
NM_004004 [GenBank, National Center for Biotechnology Information
(NCBI), www.ncbi.nlm.nih.gov/nuccore/NM_004004],
ENSG00000091137 (Ensembl, European Bioinformatics Institute,
www.ensembl.org/Homo_sapiens/Gene/Sequence?g=ENSG00000091137)
and the Cambridge reference sequence, NC_001807 (GenBank, NCBI,
www.ncbi.nlm.nih.gov/nuccore/NC_001807.4),
respectively.
Results
Genetic sequencing of 117 deaf
patients
In the present study, genetic detection was
performed in 117 deaf patients, of which 36 cases were confirmed to
carry two pathogenic mutations. The positive rate was 30.77%
(Table II), including 19 cases
with GJB2 gene mutations (16.24%), 12 cases with SLC26A4 gene
mutations (10.26%) and five cases with the 1,555A>G allelic
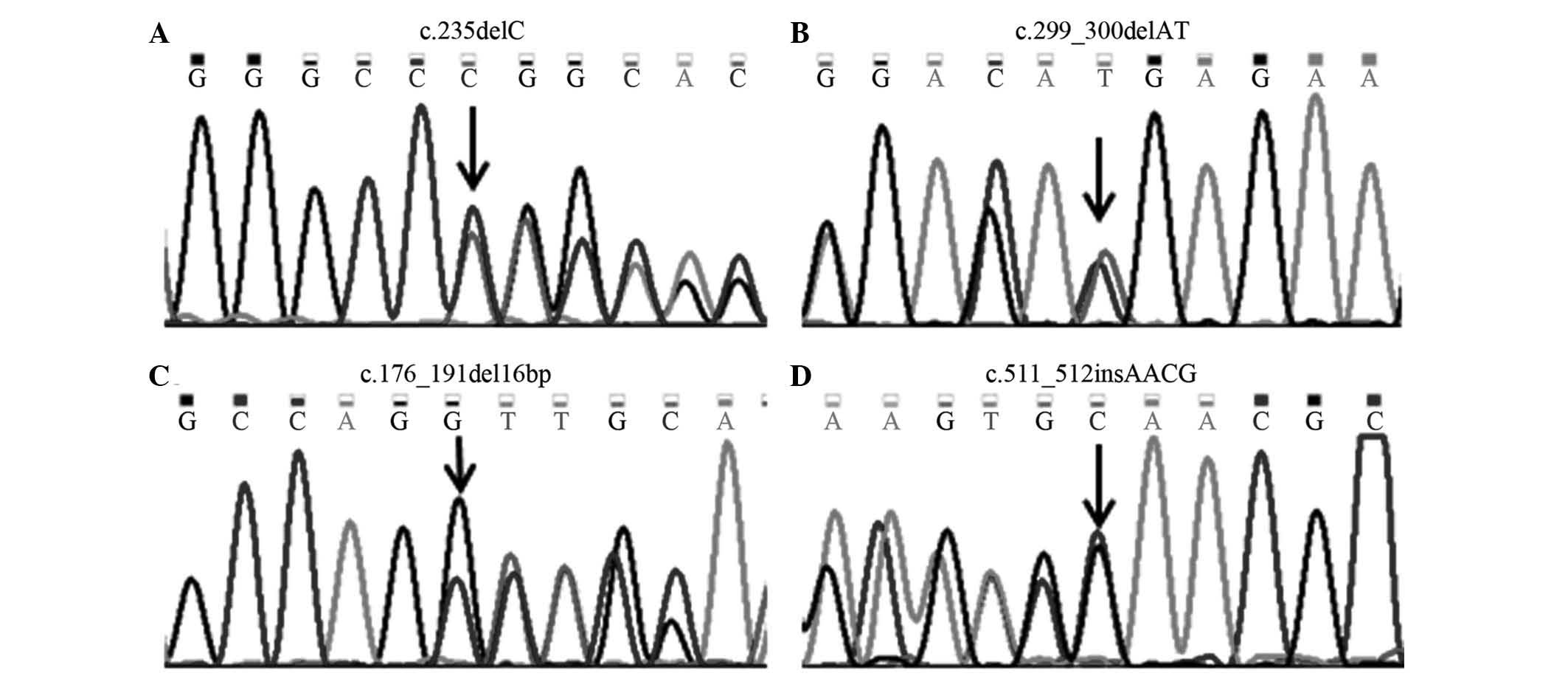
mutation of mitochondrial DNA 12S rRNA (4.27%). In total, four GJB2
gene mutations (Fig. 1), and eight
SLC26A4 gene mutations (Fig. 2)
were identified.
| Table IIMutant alleles of deafness-associated
genes in 36 deaf patients. |
Table II
Mutant alleles of deafness-associated
genes in 36 deaf patients.
| Gene | Mutant allele | Patients (n) | Total (n) | Rate (%) |
|---|
| GJB2 | 235delC/235delC | 9 | 19 | 16.24 |
|
235delC/299_300delAT | 5 | | |
|
235delC/176_191del16bp | 2 | | |
|
299_300delAT/299_300delAT | 1 | | |
|
176_191del16bp/176_191del16bp | 1 | | |
|
235delC/511_512insAACG | 1 | | |
| SLC26A4 |
IVS7-2A>G/IVS7-2A>G | 4 | 12 | 10.26 |
| IVS7-2A>G/1226
G>A | 1 | | |
|
IVS7-2A>G/1174A>T | 1 | | |
|
IVS7-2A>G/2027T>A | 1 | | |
| IVS7-2A>G/2168
A>G | 1 | | |
|
589G>A/589G>A | 1 | | |
| 1174A>T/1975
G>C | 1 | | |
|
1226G>A/1226G>A | 1 | | |
|
1079C>T/2168A>G | 1 | | |
| Mitochondrial
DNA | 1555A>G | 5 | 5 | 4.27 |
| 12S rRNA | | | | |
| Total | | | 36 | 30.77 |
Genetic sequencing of 39 deaf
couples
The 117 deaf patients recruited in the present study
included 39 pairs of deaf couples, of which four couples were
confirmed to have two deafness-causing mutations (10.26%). Among
these, deafness in one couple was caused by SLC26A4 gene mutations,
whose children will be deaf. In one deaf couple, deafness was
caused by GJB2 and SLC26A4 gene mutations, respectively. The
present study hypothesized that their children will have a low risk
of deafness with no need for prenatal diagnosis as the parents
pathogenic mutations are in different genes, the child will be
heterozygous for these two genes which are associated with
autosomal recessive hearing loss. Furthermore, in one couple, the
husband had two GJB2 gene mutations, whereas the wife had
1,555A>G mutation of mitochondrial DNA 12S rRNA. Their children
would carry a GJB2 gene heterozygous mutation and mitochondrial DNA
mutation, and may be advised to avoid taking aminoglycoside
antibiotics as the mutation 1,555A>G is important in
aminoglycoside-induced nonsyndromic deafness (18). In addition, the husband of one
couple had 1,555A>G mutation of mitochondrial DNA 12S rRNA,
whereas the wife carried two GJB2 gene mutation. The present study
hypothesized that their children would be at low risk of deafness
due to their child carrying a heterozygous GJB2 gene mutation. The
results of genetic sequencing indicated that, in 11 couples, only
one of each couple (28.21%) was confirmed to have deafness-causing
mutations, which indicated that their offspring were at low risk of
deafness. Table III shows the
results of genetic analyses.
| Table IIIGenetic sequencing of 15 couples. |
Table III
Genetic sequencing of 15 couples.
| Couple | Gender/age
(years) | GJB2 mutation | SLC26A4
mutation | 12S rRNA
mutation |
|---|
| 1 | Female/20 |
299_300delAT/299_300delAT | – | – |
| Male/28 | – | – | – | |
| 2 | Female/29 |
235delC/511_512insAACG | – | – |
| Male/30 | – | – | – | |
| 3 | Female/22 | – | – | – | |
| Male/29 | | – | – | 1555A>G |
| 4 | Female/28 | | – | – | 1555A>G |
| Male/29 |
235delC/235delC | – | – |
| 5 | Female/24 | | – |
1226G>A/1226G>A | – |
| Male/25 | | – |
IVS7-2A>G/2027T>A | – |
| 6 | Female/24 | | – | – | 1555A>G |
| Male/24 | – | – | – | |
| 7 | Female/28 |
235delC/235delC | – | – |
| Male/28 | – | – | – | |
| 8 | Female/23 |
235delC/235delC | – | – |
| Male/24 | | – | – | – |
| 9 | Female/26 | | – | – | – |
| Male/27 | | – | IVS7-2A>G/1226
G>A | – |
| 10 | Female/28 | | – | – | – |
| Male/30 | | – | | 1555A>G |
| 11 | Female/25 | |
235delC/176_191del16bp | – | – |
| Male/25 | | – | – | – |
| 12 | Female/25 | |
235delC/235delC | – | – |
| Male/26 | | – | – | – |
| 13 | Female/26 | | – | – | |
| Male/31 | | – |
589G>A/589G>A | – |
| 14 | Female/28 | |
235delC/299_300delAT | – | – |
| Male/29 | | – |
IVS7-2A>G/IVS7-2A>G | – |
| 15 | Female/29 | |
235delC/176_191del16bp | – | – |
| Male/34 | | – | – | 1555A>G |
Genetic sequencing of families with deaf
children and prenatal diagnosis
In the 39 deaf patients, 17 cases were identified
with GJB2 gene or SLC26A4 gene mutations. Subsequently, their
parents received genetic analysis to confirm whether they were
carriers (Table IV). In 7/17
families, prenatal diagnosis was performed during their next
pregnancy. The results showed that two fetuses had the same
genotype as the proband. These two families decided to terminate
the pregnancy. No mutations were identified in one fetus, and the
genotypes of the other four fetuses were the same as one parent.
The result of clinical follow-up demonstrated that they had normal
hearing following birth.
| Table IVGenetic sequencing of 17 couples with
deaf children. |
Table IV
Genetic sequencing of 17 couples with
deaf children.
| Couple | Mutation
| Follow-up |
|---|
| Child
(proband) | Father | Mother | Fetus |
|---|
| 1a |
235delC/299_300delAT | 299_300delAT | 235delC |
235delC/299_300delAT | Ending
pregnancy |
| 2a |
235delC/299_300delAT | 235delC | 299_300delAT |
299_300delAT/WT | Normal hearing |
| 3a |
235delC/235delC | 235delC | 235delC | 235delC/WT | Normal hearing |
| 4a |
176_191del16bp/176_191del16bp | 176_191del16bp | 176_191del16bp |
176_191del16bp/WT | Normal hearing |
| 5b |
IVS7-2A>G/IVS7-2A>G | IVS7-2A>G | IVS7-2A>G | WT/WT | Normal hearing |
| 6b |
IVS7-2A>G/IVS7-2A>G | IVS7-2A>G | IVS7-2A>G |
IVS7-2A>G/IVS7-2A>G | Ending
pregnancy |
| 7b |
IVS7-2A>G/2168A>G | IVS7-2A>G | 2168A>G |
IVS7-2A>G/WT | Normal hearing |
| 8a |
235delC/299_300delAT | 299_300delAT | 235delC | | |
| 9a |
235delC/299_300delAT | 299_300delAT | 235delC | | |
| 10a |
235delC/235delC | 235delC | 235delC | | |
| 11a |
235delC/235delC | 235delC | 235delC | | |
| 12a |
235delC/235delC | 235delC | 235delC | | |
| 13a |
235delC/235delC | 235delC | 235delC | | |
| 14b |
IVS7-2A>G/1174A>T | 1174A>T | IVS7-2A>G | | |
| 15b |
IVS7-2A>G/IVS7-2A>G | IVS7-2A>G | IVS7-2A>G | | |
| 16b |
1174A>T/1975G>C | 1174A>T | 1975G>C | | |
| 17b |
1079C>T/2168A>G | 2168A>G | 1079C>T | | |
Discussion
The provision of appropriate genetic counseling for
patients with hearing loss remains a challenge in clinical
practice. Deafness exhibits marked genetic heterogeneity and
phenotypic variability (1,2). At present, hundreds of genes have
been identified to cause hereditary hearing loss, including the
GJB2 gene, SLC26A4 gene and the mitochondrial DNA 12S rRNA
1,555A>G mutation, which have been confirmed to be closely
associated with NSHL (1,16–23).
The present study used Sanger sequencing to analyze
mutations of GJB2, SLC26A4 and the mitochondrial DNA 12S rRNA
1,555A>G in 117 patients with NSHL. The results revealed that 36
of the 117 patients (30.77%) carried two deafness-causing
mutations, including the GJB2 gene mutations (16.24%), SLC26A4 gene
mutations (10.26%) and mitochondrial DNA 12SrRNA 1555 locus
mutation (4.27%). The 235delC mutation in the GJB2 gene mutation
has been identified at the highest rate in patients in the present
study and is also the most common in the Asian population (24). The present study identified the
mitochondrial DNA 12S rRNA 1,555A>G locus mutation in ~4.27% of
the patients, which follows the pattern of maternal inheritance
(11,18). It may be the situation that there
are healthy individuals with normal hearing carrying the
1,555A>G mutation. Thus, once this mutation is identified, the
family members can avoid the use of aminoglycoside antibiotics,
preventing drug-induced deafness (25). The SLC26A4 gene contains 21 exons.
The present study selected certain hot-spot regions, SLC26A4 exons
5, 7, 8, 10, 17 and 19, for sequencing. The IVS7-2A>G mutation
is the most common mutation in the SLC26A4 gene (10). For patients with one allelic
variant in the hot-spot regions, the remaining exons were sequenced
one by one until two mutant alleles had been identified. The
results found the second mutant allele, the missense mutation
1079C>T, in exon 9 of one patient. In addition, 11 patients
carrying SLC26A4 mutations were detected using hot-spot region
screening, whereas only one case was identified by complete exon
sequencing. According to the SLC26A4 gene mutation spectrum, the
Sanger sequencing method detecting hot-spot regions of the SLC26A4
gene has been found to have clinical value in identifying mutations
associated with deafness (14).
In the present study, there were 39 deaf couples.
Traditionally, it is thought that there is a high possibility of
deaf couples having hearing-impaired offspring, however, the
majority of couples with hearing impairment have deafness induced
by environmental causes or due to different pathogenic genes, thus,
the risk of their children being deaf is low (12). Additionally, in couples with
deafness due to GJB2 and SLC26A4 gene mutation, their children were
predicted to be carriers with normal hearing. In one couple, in
which the mother had hearing loss caused by the mitochondrial DNA,
12S rRNA mutation, their children were predicted to be a carrier
with normal hearing, in the absence of use with ototoxic drugs.
However, they cannot be a carrier of the mutational gene if the
father has hearing loss caused by the mitochondrial DNA 12S rRNA
mutation, the children would be predicted to have normal hearing.
Therefore, genetic analysis of deafness-associated genes can reduce
anxiety in deaf couples, and determine the risk in their next
pregnancy.
In the present study, 3 deafness-associated genes in
39 deaf patients and their parents were sequenced, and the results
revealed 17 cases carried two confirmed pathogenic mutations, whose
parents were both carriers of the same genetic mutation. In
addition, seven pregnant women were offered prenatal diagnosis. The
genetic analysis revealed that the genotypes of two fetuses were
the same as the probands. Of the remaining fetuses, four were
carriers, and one was identified without mutations in the genes
analyzed.
In conclusion, the gene sequencing performed in the
present study was convenient and reliable for the genetic testing
of NSHL. In addition, the method could be applied in clinical
laboratories due to its simple process and low cost, providing
easier genetic assessment. In addition, results from the present
study provided accurate information for genetic counseling and
prenatal diagnosis.
Acknowledgments
The present study was supported by the Program for
Leading Talents and Medical Innovative Team of Jiangsu Province
(grant no. LJ201109), the Special Funds of Clinical Medicine
Science and Technology of Jiangsu Province (grant no. BL2012039),
the Science and Technology Research and Development Program of
Health Bureau of Nanjing City (grant no. YKK11059) and the Training
Program for Young Talents of Nanjing City.
References
|
1
|
Shearer AE and Smith RJ: Genetics:
Advances in genetic testing for deafness. Curr Opin Pediatr.
24:679–686. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cryns K and Van Camp G: Deafness genes and
their diagnostic applications. Audiol Neurootol. 9:2–22. 2004.
View Article : Google Scholar
|
|
3
|
Hoefsloot LH, Feenstra I, Kunst HP and
Kremer H: Genotype phenotype correlations for hearing impairment:
Approaches to management. Clin Genet. 85:514–523. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rabionet R, Gasparini P and Estivill X:
Molecular genetics of hearing impairment due to mutations in gap
junction genes encoding beta connexins. Hum Mutat. 16:190–202.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kenneson A, Van Naarden Braun K and Boyle
C: GJB2 (connexin 26) variants and nonsyndromic sensorineural
hearing loss: A HuGE review. Genet Med. 4:258–274. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Snoeckx RL, Huygen PL, Feldmann D, Marlin
S, Denoyelle F, Waligora J, Mueller-Malesinska M, Pollak A, Ploski
R, Murgia A, et al: GJB2 mutations and degree of hearing loss: A
multicenter study. Am J Hum Genet. 77:945–957. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ohtsuka A, Yuge I, Kimura S, Namba A, Abe
S, Van Laer L, Van Camp G and Usami S: GJB2 deafness gene shows a
specific spectrum of mutations in Japan, including a frequent
founder mutation. Hum Genet. 112:329–333. 2003.PubMed/NCBI
|
|
8
|
Hilgert N, Smith RJ and Van Camp G:
Forty-six genes causing nonsyndromic hearing impairment: Which ones
should be analyzed in DNA diagnostics? Mutat Res. 681:189–196.
2009. View Article : Google Scholar
|
|
9
|
Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H,
Zong L, Guan J, Xu BC, Wang DY, Han MK, et al: A distinct spectrum
of SLC26A4 mutations in patients with enlarged vestibular aqueduct
in China. Clin Genet. 72:245–254. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du W, Guo Y, Wang C, Wang Y and Liu X: A
systematic review and meta-analysis of common mutations of SLC26A4
gene in Asian populations. Int J Pediatr Otorhinolaryngol.
77:1670–1676. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kokotas H, Petersen MB and Willems PJ:
Mitochondrial deafness. Clin Genet. 71:379–391. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yuan Y, You Y, Huang D, Cui J, Wang Y,
Wang Q, Yu F, Kang D, Yuan H, Han D and Dai P: Comprehensive
molecular etiology analysis of nonsyndromic hearing impairment from
typical areas in China. J Transl Med. 7:792009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai P, Yu F, Han B, Liu X, Wang G, Li Q,
Yuan Y, Liu X, Huang D, Kang D, et al: GJB2 mutation spectrum in
2,063 Chinese patients with nonsyndromic hearing impairment. J
Transl Med. 7:262009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan Y, Guo W, Tang J, Zhang G, Wang G,
Han M, Zhang X, Yang S, He DZ and Dai P: Molecular epidemiology and
functional assessment of novel allelic variants of SLC26A4 in
non-syndromic hearing loss patients with enlarged vestibular
aqueduct in China. PLoS One. 7:e499842012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Z, Li R, Chen J, Liao Z, Zhu Y, Qian Y,
Xiong S, Heman-Ackah S, Wu J, Choo DI and Guan MX: Mutational
analysis of the mitochondrial 12S rRNA gene in Chinese pediatric
subjects with aminoglycoside-induced and non-syndromic hearing
loss. Hum Genet. 117:9–15. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Estivill X, Fortina P, Surrey S, Rabionet
R, Melchionda S, D'Agruma L, Mansfield E, Rappaport E, Govea N,
Milà M, et al: Connexin-26 mutations in sporadic and inherited
sensorineural deafness. Lancet. 351:394–398. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Morell RJ, Kim HJ, Hood LJ, Goforth L,
Friderici K, Fisher R, Van Camp G, Berlin CI, Oddoux C, Ostrer H,
et al: Mutations in the connexin 26 gene (GJB2) among Ashkenazi
Jews with nonsyndromic recessive deafness. N Engl J Med.
339:1500–1505. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Prezant TR, Agapian JV, Bohlman MC, Bu X,
Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L and Rotter JI:
Mitochondrial ribosomal RNA mutation associated with both
antibiotic-induced and non-syndromic deafness. Nat Genet.
4:289–294. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao H, Li R, Wang Q, Yan Q, Deng JH, Han
D, Bai Y, Young WY and Guan MX: Maternally inherited
aminoglycoside-induced and nonsyndromic deafness is associated with
the novel C1494T mutation in the mitochondrial 12S rRNA gene in a
large Chinese family. Am J Hum Genet. 74:139–152. 2004. View Article : Google Scholar
|
|
20
|
Pryor SP, Madeo AC, Reynolds JC, Sarlis
NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA
and Griffith AJ: SLC26A4/PDS genotype-phenotype correlation in
hearing loss with enlargement of the vestibular aqueduct (EVA):
Evidence that Pendred syndrome and non-syndromic EVA are distinct
clinical and genetic entities. J Med Genet. 42:159–165. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Q, Zhu QW, Yuan YY, Huang SS, Han DY,
Huang DL and Dai P: Identification of SLC26A4 c.919-2A>G
compound heterozygosity in hearing-impaired patients to improve
genetic counseling. J Transl Med. 10:2252012. View Article : Google Scholar :
|
|
22
|
Sun SC, Liu YX, Peng YS, Li HF and Xie CY:
Analysis of GJB2 gene coding sequence in patients with nonsyndromic
hearing loss. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 28:409–413.
2011.In Chinese. PubMed/NCBI
|
|
23
|
Dai P, Stewart AK, Chebib F, Hsu A,
Rozenfeld J, Huang D, Kang D, Lip V, Fang H, Shao H, et al:
Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S.
patients with nonsyndromic hearing loss. Physiol Genomics.
38:281–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chun JY, Shin SK, Min KT, Cho W, Kim J,
Kim SO and Hong SP: Performance evaluation of the TheraTyper-GJB2
assay for detection of GJB2 gene mutations. J Mol Diagn.
16:573–583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kent A, Turner MA, Sharland M and Heath
PT: Aminoglycoside toxicity in neonates: Something to worry about?
Expert Rev Anti Infect Ther. 12:319–331. 2014. View Article : Google Scholar : PubMed/NCBI
|