Introduction
Osteoarthritis (OA) is a common, predominantly
age-related joint disorder, which is characterized by the loss of
chondrocytes, degradation of the extracellular matrix (ECM),
subchondral bone remodeling and synovial inflammation (1). It has been demonstrated that
chondrocyte apoptosis is associated with the initiation and
severity of articular cartilage degradation (2), and chondrocyte apoptosis increases in
human OA cartilage (3).
Chondrocyte apoptosis is predominantly responsible for the
progressive cartilage degradation, which occurs in osteoarthritis
(OA) and has gradually become one of the potential therapeutic
targets for OA (4).
Mitogen-activated protein kinase (MAPK) is a
serine/threonine kinase, the signaling cascades of which control
complex programs, including proliferation, differentiation and
apoptosis (5,6). There are three major classes of MAPKs
in mammals, the extracellular signal-regulated kinases (ERKs),
c-jun N-terminal kinase and p38 (7). Chondrocyte apoptosis in experimental
OA is regulated by the p38/MAPK signaling pathway (8), and the p38/MAPK signal transduction
pathway has also been implicated as a critical factor in nitric
oxide (NO)-induced articular chondrocyte apoptosis in rabbits
(9). The p38/MAPK cascades can be
activated or phosphorylated by a series of inflammatory factors and
stimuli from the environment, and phosphorylated (p)-p38 is then
shuttled into the nucleus, where it activates transcription
factors, initiates the expression of upstream apoptosis-associated
genes and promotes the apoptosis of chondrocytes (10,11).
Caspase-3 is a key member of the caspase family, which can cleave
DNA repair proteins, including poly (ADP-ribose) polymerase,
cytoskeletal proteins and inhibitor of caspase-activated
deoxytribonuclease, thereby leading to the fragmentation of DNA and
eventual cell apoptosis (12). NO
is an important physiological and pathological signaling molecule,
which can be produced by inflammatory stimuli, including sodium
nitroprusside (SNP). It can inhibit the proliferation of
chondrocytes and induce chondrocyte apoptosis, thereby damaging
cartilage and impairing joint cartilage repair (13). NO can induce chondrocyte apoptosis
through multiple pathways, including the p38/MAPK pathway.
Inhibiting p38 signaling may effectively inhibit these negative
effects, prevent chondrocyte apoptosis and promote cartilage
repair.
Carboxymethylated chitosan (CMCS) is a soluble
derivative of chitosan, which possesses several desirable
physiochemical and biological features. Our previous studies showed
that CMCS significantly suppresses the degeneration of cartilage in
OA and protects chondrocytes from interleukin-1β-induced apoptosis
(14,15). It has been found that CMCS can
stimulate the proliferation and synthesis of nerve growth factor in
Schwann cells (SCs) by activation of the MAPK kinase (MEK)/ERK,
phosphatidylinositol 3-kinase/Akt and Wnt/β-catenin signaling
pathways (16,17), and protects SCs and nucleus
pulposus cells from hydrogen peroxide-induced apoptosis (18,19).
However, whether CMCS can inhibit chondrocyte apoptosis through
inhibiting p38/MAPK signaling remains to be elucidated.
The aim of the present study was to investigate
whether CMCS can inhibit chondrocyte apoptosis using an in
vitro model of rat articular chondrocyte apoptosis. Numerous
studies have indicated that the p38/MAPK signaling pathway is one
of the most important signaling pathways in NO-induced cell
apoptosis, including chondrocytes (9). Therefore, the present study also
aimed to elucidate the mechanism underlying the protective role of
CMCS on chondrocytes by examining activation of the p38/MAPK
signaling pathway.
Materials and methods
Animals and reagents
A total of 24 healthy Sprague-Dawley (SD) rats (3
weeks old) with an average body weight of 362±35 g were obtained
from the Experimental Animal Center of Wuhan University (Wuhan,
China). Dulbecco's modified Eagle's medium (DMEM) was obtained from
Gibco (Thermo Fisher Scientific, Inc., Waltham, MA, USA), fetal
bovine serum (FBS) was obtained from GE Healthcare Life Sciences
(Logan, UT, USA). CMCS (purity >99%) was supplied by the
Institute of Chemistry and Environmental Science of Wuhan
University (Wuhan, China). Primers were synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). Anti-p38 and anti-p-p38
antibodies were obtained from Cell Signaling Technology, Inc.
(Beverly, MA, USA). Anti-β-actin antibody was obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). A Caspase-3
Activity Assay kit (cat. no. C1115) was purchased from the Beyotime
Institute of Biotechnology (Haimen, China). A Cell Counting Kit-8
(CCK-8; cat. no. CK04) was purchased from Dojindo Molecular
Technologies, Inc. (Kumamoto, Japan). SB203580 (cat. no. S3807),
type-2 collagenase (cat. no. C6885) and SNP (cat. no. 1614501) were
obtained from Sigma-Aldrich (St. Louis, MO, USA). An Annexin
V-Fluorescein isothiocyanate (FITC)/propidium iodide (PI) Apoptosis
Detection kit (cat. no. 556547) was obtained from BD Biosciences
(Franklin Lakes, NJ, USA). Other commonly used reagents were of
high purity and commercially available.
Cell isolation and culture
Rat articular chondrocytes were isolated and
cultured, as previously described (20). The utilization of rat articular
cartilage was approved by the Animal Ethics Committee of Wuhan
University. Rats were anesthetized with a single intraperitoneal
injection of 1% sodium pentobarbital (40 mg/kg; purchased from
Sigma-Aldrich; cat. no. 1507002). The articular cartilage tissue
was removed from the knee joints of the 3-week-old rats using a
scalpel. The extracted cartilage was cut into 1×1×1 mm3
sections. Animals were sacrificed by an overdose of sodium
pentobarbital. Subsequently, chondrocytes were isolated from the
extracted cartilage by enzymatic digestion with 0.25% trypsin
(Sigma-Aldrich) in phosphate-buffered saline (PBS) for 1 h, and
0.2% type-2 collagenase (381 U/mg; Sigma-Aldrich) in DMEM for 4 h.
Following collection of individual cells by brief centrifugation,
the chondrocytes were resuspended in DMEM supplemented with 10%
FBS, 100 U/ml streptomycin and 100 U/ml penicillin (Hyclone, Logan,
UT, USA). The culture medium was replaced every other day, and the
second and third passage of chondrocytes were used in the
subsequent experiments.
Cell treatment and grouping
To establish the apoptotic model of cultured
chondrocytes, SNP was used, which generates NO (21). Briefly, the chondrocytes
(5×104 cells/cm2) were seeded and cultured at
37°C for 24 h, following which the cells were randomly divided into
five groups, in which the medium was replaced with complete medium
(DMEM with 10% FBS and antibiotics) containing 0, 0.5, 1, 2 or 3 mM
SNP, cultured at 37°C for different durations (3, 6, 12 and 24 h).
CMCS was added into the culture medium at different concentrations
(50, 100 and 200 µg/ml). Subsequently, in order to determine
the activation of the p38/MAPK signaling pathway, the p38/MAPK
specific inhibitor, SB203580 was used (22).
Cell viability assay
The chondrocytes were cultured in 96-well
microplates at a density of 2×104 cells per well with
DMEM containing 0.1% FBS at 37°C for 24 h, and then exposed to
different concentrations of SNP (0.5, 1, 2 and 3 mM), CMCS (50, 100
and 200 µg/ml) or SB203580 (10 µM) for 24 h, and for
different durations (0, 3, 6, 12 or 24 h). For the quantitative
analysis of the cell viability, 10 µl CCK-8 solution was
added to each well and incubated at 37°C for 1 h. The optical
densities were measured at 450 nm using an ELx800 Absorbance
Microplate reader (BioTek Instruments, Inc., Winooski, VT, USA).
Cell viability was determined as a percentage of the number of
control (untreated) cells. Viability in the control group was
designated as 100%, and cell viability in the treatment groups was
determined as follows:
Viability (% of control) = [(Ae − Ab) / (Ac − Ab)] ×
100%. Ae, Ab and Ac represent the A450 of the experimental, blank
and control groups, respectively. All experiments were performed in
triplicate in three independent experiments.
Flow cytometric analysis
The apoptotic rates of the chondrocytes was measured
using flow cytometry (FCM), according to the manufacturer's
protocol of the Annexin V-FITC/PI Apoptosis Detection kit. In
brief, the chondrocytes were treated with SNP (0.5, 1, 2 and 3 mM),
CMCS (50, 100 and 200 µg/ml) or SB203580 (10 µM).
Following culture of the chondrocytes for the indicated durations
(0, 3, 6, 12 or 24 h), trypsinization was performed with 0.25%
trypsin without ethylene diamine tetraacetic acid for analysis of
apoptosis using the Annexin V-FITC/PI Apoptosis Detection kit. Cell
apoptosis was analyzed using a BD FACSVerse™ flow cytometer (Becton
Dickinson, Heidelberg, Germany) at 488 nm. The data were analyzed
using CellQuest™ software (version 4.01; Becton Dickinson).
Caspase-3 activity assay
Caspase-3 activity was determined using a
colorimetric assay kit (cat. no. C1115; Beyotime Institute of
Biotechnology). The chondrocytes were first cultured in a mixture
of DMEM supplemented with 5% FBS, SNP and CMCS. Following
incubation at 37°C for 24 h, the cells were treated, according to
the manufacturer's protocol. The cell lysates were prepared, and
assays were performed in 96-well plates (at a density of
2×104 cells/well) by incubating 10 µl cell lysate
per sample in 80 µl reaction buffer (1% NP-40, 20 mM
Tris-HCl, 137 mM NAD and 10% glycerol) containing 10 µl
caspase-3 substrate (Ac-DEVD-pNA; Beyotime Institute of
Biotechnology). The cell lysates were incubated at 37°C for 4 h.
The absorbance at 405 nm was read using an ELx800 Absorbance
Microplate reader (BioTek Instruments, Inc.). Caspase-3 activities
were determined as the percentage of enzyme activity, compared with
the control. All experiments were performed in triplicate.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The mRNA of the treated chondrocytes was extracted
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. An ABI Prism 7500
real-time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) was used to detect the mRNA expression levels of the target
genes. The sequences of the primers used, for B cell lymphoma
(Bcl)-2, Bcl-2-associated X protein (Bax) and GAPDH, are shown in
Table I.
 | Table IPrimer sequences of target genes. |
Table I
Primer sequences of target genes.
| Gene | Primer
sequence | Product size
(bp) |
|---|
| Bcl-2 | F:
5′-GCGTCAACAGGGAGATGTCA-3′ | 225 |
| R:
5′-GGTATGCACCCAGAGTGATG-3′ | |
| Bax | F:
5′-GGCGAATTGGAGATGAACTG-3′ | 209 |
| R:
5′-GATCAGCTCGGGCACTTTAG-3′ | |
| GAPDH | F:
5′-TGTCTCCTGCGACTTCAACAG-3′ | 256 |
| R:
5′-GAGGCCATGTAGGCCATGAG-3′ | |
For RT reaction, 5 µl mRNA was reverse
transcribed into cDNA and amplified using specific reverse primers
of the target genes using a RevertAid™ First Strand cDNA Synthesis
kit (Fermentas; Thermo Fisher Scientific, Inc.). The PCR reaction
mixture included 1 µl forward and reverse primers (10
µM), 1 µl SYBR Green I fluorescent dye and 1
µl cDNA. The PCR conditions were as follows: Initial step at
95°C for 2 min, followed by 40 cycles at 95°C for 20 sec and 60°C
for 40 sec. The relative expression levels of target genes were
analyzed using the comparative cycle threshold method (23). Data were standardized by GAPDH. The
results were obtained from three independent experiments.
Western blot analysis
The chondrocytes were treated with SNP, with or
without CMCS, following which the chondrocytes were lysed in lysis
buffer at 4°C for 30 min. The lysates were centrifuged at 13,000 g
or 15 min at 4°C. The supernatants were collected and stored at
−80°C. Protein concentrations were determined using a bicinchoninic
acid method (Pierce Biotechnology; Thermo Fisher Scientific, Inc.).
Total protein (50 µg) was separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (Beyotime Institute of
Biotechnology) and electroblotted onto polyvinylidene difluoride
membrane (EMD Millipore, Billerica, MA, USA). After being blocked
with 5% non-fat milk in Tris-buffered saline, the membrane was
incubated overnight at 4°C with primary antibodies against p38
(rabbit polyclonal IgG, cat. no. 9212; Cell Signaling Technology,
Inc.), p-p38 (rabbit polyclonal IgG, cat. no. 9211; Cell Signaling
Technology, Inc.) and β-actin (rabbit polyclonal IgG, cat. no.
sc-10731; Santa Cruz Biotechnology, Inc.) at dilutions of 1:200 for
2 h at room temperature. Subsequently, the membrane was incubated
with a peroxidase-conjugated secondary antibody (goat anti-rabbit
horseradish peroxidase-conjugated polyclonal IgG, cat. no.
sc-45101; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. Signals were detected by enhanced chemiluminescence.
Optical band density was quantified using a Geliance 200 Imaging
system (PerkinElmer, Waltham, MA, USA) and Gene Snap software
(version 6.08.04; Syngene, Cambridge, UK). The values for p38 and
p-p38 were normalized to that of β-actin.
Statistical analysis
The data are presented as the mean ± standard
deviation. Statistical significance was analyzed by one-way
analysis of variance using SPSS, version 16.0 (SPSS, Inc., Chicago,
IL, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
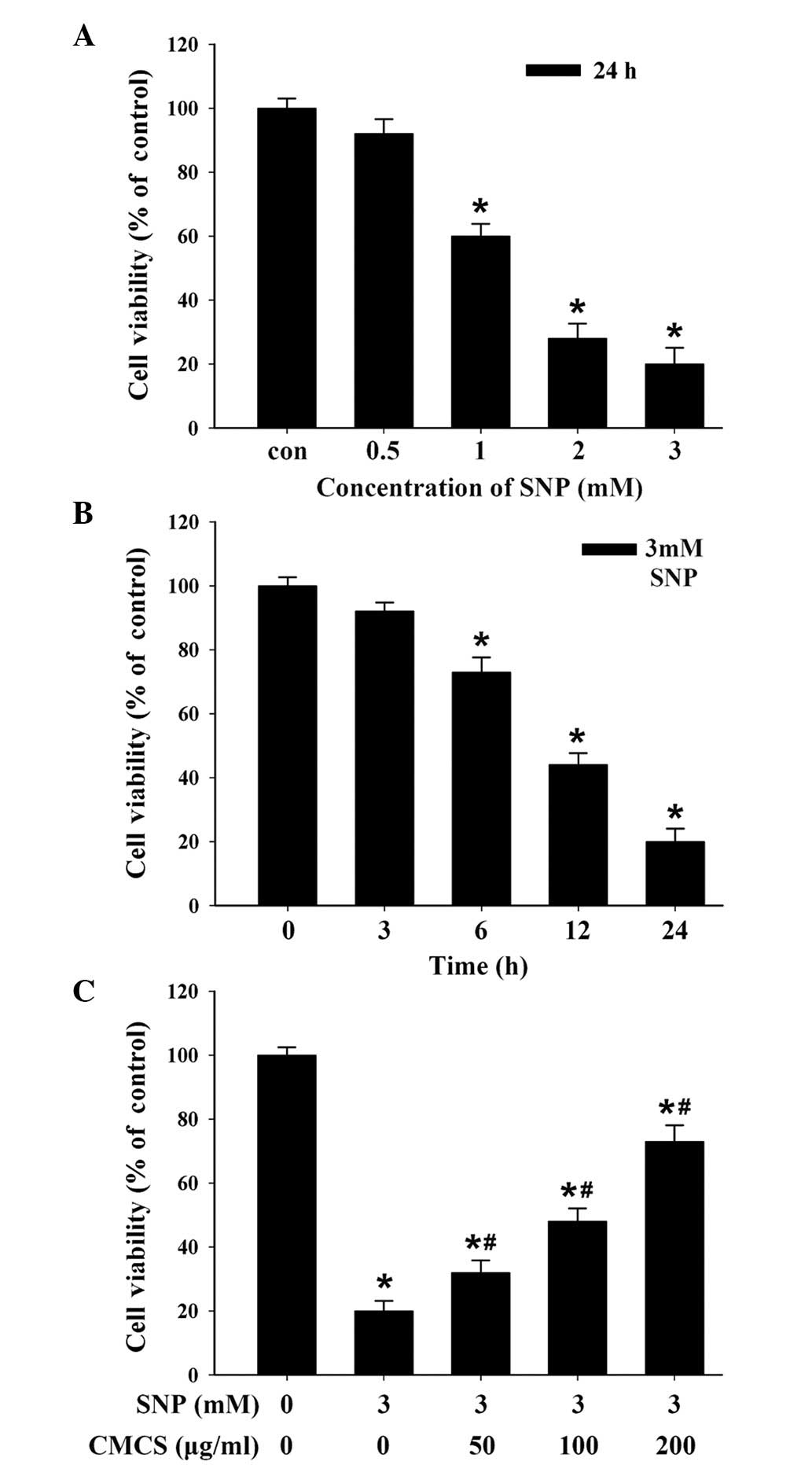
CMCS increases cell viability in
SNP-induced chondrocytes
To determine the protective effects of CMCS on
chondrocytes, a model of apoptosis was established in the present
study by exposure of chondrocytes to SNP, an inorganic compound
with the formula
Na2[Fe(CN)5-NO]·2H2O. As shown in
Fig. 1A, SNP induced a
dose-dependent cytotoxic effect on the chondrocytes, and treatment
with 3 mM SNP for 24 h induced a decrease in cell viability by
>90%. In addition, treatment with 3 mM SNP for different
durations induced a time-dependent cytotoxic effect (Fig. 1B). Therefore, the results indicated
that SNP induced dose- and time-dependent cytotoxic effects on the
chondrocytes. Based on these data, chondrocytes treated with 3 mM
SNP for 24 h were used as the in vitro apoptosis model for
subsequent experiments. To investigate the protective effects of
CMCS on chondrocytes, the chondrocytes were pretreated with
different concentrations of CMCS for 24 h prior to SNP induction,
following which cell viability was measured. As shown in Fig. 1C, CMCS caused an increase in
SNP-induced cytotoxicity, indicating that CMCS protected the
chondrocytes from the SNP-induced decrease in viability, and may
offer potential as a potent drug to treat OA.
CMCS inhibits SNP-induced apoptosis in
rat chondrocytes
To confirm the apoptosis-inducing effects of SNP,
Annexin V-FITC/PI staining, followed by FCM was performed. As shown
in Fig. 2, the numbers of normal
cells in the SNP-induced groups were significantly lower, compared
with the number in the control group, whereas the numbers of early
apoptotic, late apoptotic and dead cells were significantly higher
in number, compared with those in the control group. Increasing
concentrations and durations increased the apoptotic rate, and the
maximum pro-apoptotic effect was observed following treatment with
3 mM SNP for 24 h. These results are consistent with previous
studies, demonstrating that SNP treatment leads to chondrocyte
apoptosis (24,25). Following treatment with different
concentrations of CMCS, the proportion of early apoptotic, late
apoptotic and dead cells decreased, and there was a dose-dependent
effect of CMCS treatment, with 200 µg/ml CMCS exerting the
most marked inhibitory effect on SNP-induced apoptosis.
CMCS inhibits the activation of p38 and
decreases caspase-3 activity
To further examine the mechanism underlying the
effects of CMCS on signaling cascades, the present study analyzed
the activation of p38/MAPK. As shown in Fig. 3, quantification of the Western blot
bands showed that the protein levels of p-p38 were significantly
higher in the SNP-treated groups, compared with the control group.
Following treatment with CMCS or the p38 inhibitor, SB203580, the
expression of p-p38 was decreased. As shown in Fig. 4, caspase-3 activity was
significantly increased, to 63% of the control, in the SNP-induced
chondrocytes, however, pretreatment with 50, 100 or 200
µg/ml CMCS attenuated the SNP-induced increase in caspase-3
activity by ~41, 54 and 56%, respectively.
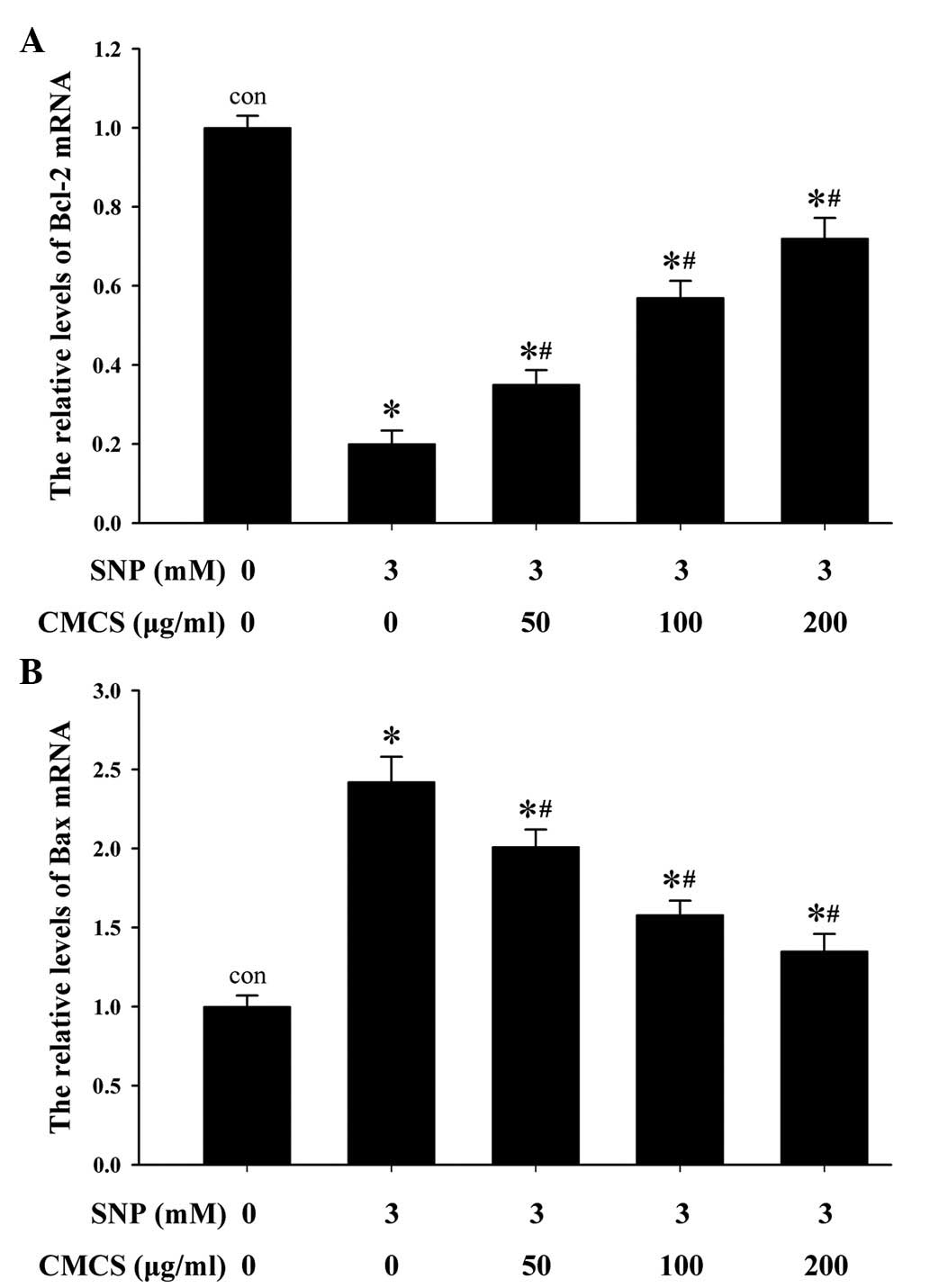
CMCS increases the expression of Bcl-2
and decreases the expression of Bax in SNP-induced
chondrocytes
In order to further investigate the mechanisms
underlying the effects of CMCS on SNP-induced chondrocyte
apoptosis, the mRNA expression levels of pro-apoptotic Bax and
anti-apoptotic Bcl-2 were detected using RT-qPCR analysis. As shown
in Fig. 5A, the mRNA expression
levels of Bcl-2 were markedly elevated in the CMCS-treated
chondrocytes, compared with the 3 mM SNP-treated group without
CMCS, at CMCS concentrations of 50 µg/ml (1.68±0.16-fold),
100 µg/ml (2.23±0.21-fold) and 200 µg/ml
(4.12±0.23-fold). As shown in Fig.
5B, 3 mM SNP significantly
increased the mRNA level of Bax (2.14±0.17-fold), compared with the
control group. This increase was reversed by the addition of CMCS.
These findings indicated that CMCS inhibited the mRNA expression of
Bax and promoted the mRNA expression of Bcl-2 in the SNP-induced
chondrocytes.
CMCS prevents SNP-induced apoptosis via
p38/MAPK
As p38/MAPK was found to be activated in response to
SNP., the present study proceeded to address the role of p38/MAPK
in the CMCS-induced inhibition of chondrocyte apoptosis, by
treating the chondrocytes with SB203580, a specific inhibitor of
p38 activation. The chondrocytes were pretreated with 10 µM
SB203580 for 1 h and then stimulated with CMCS for 24 h. A
subsequent CCK-8 assay showed that pretreatment with CMCS and/or
SB203580 decreased the inhibitory effect of SNP on viability
(Fig. 6A). In addition, FCM showed
that pretreatment with CMCS or SB203580 prevented SNP-induced
apoptosis (Fig. 6B). These
findings suggested that CMCS prevented NO-induced apoptosis,
possibly through the inhibition of p38/MAPK activation.
Discussion
CMCS has shown therapeutic effects on non-alcoholic
fatty liver disease and hydrophobic drug delivery (26,27).
Our previous study indicated that CMCS inhibits chondrocyte, SC and
nucleus pulposus cell apoptosis in vitro (15,18,19).
However, the mechanism or signaling pathway underlying the effect
of CMCS in OA remains to be fully elucidated. The results of the
present study indicated that pre-treatment with CMCS in SNP-induced
chondrocytes promoted the expression of Bcl-2, inhibited the
expression of Bax and reduced caspase-3 activation. These effects
by CMCS are partially mediated via the inhibition of p38/MAPK
signaling.
OA is a degenerative joint disease with multiple
under-lying pathogenic mechanisms, caused by various risk factors,
including NO (28),
polychlorinated biphenyl 126 (29), bupivacaine and levobupivacaine
(30), and C/EBP homologous
protein (31) has been reported to
induce apoptosis in chondrocytes. As these apoptosis-inducing
substances may be involved in the pathogenesis of OA, manipulation
of the mechanism mediated by these stimuli has the potential for
substantial therapeutic effects.
The loss of chondrocyte function, ECM degradation
and apoptosis are crucial in the progression of OA, thus inhibiting
the development of these factors is important for the treatment of
OA. To the best of our knowledge, the signaling pathways involved
in the effects of CMCS on OA remain to be fully elucidated. In the
present study, the anti-apoptotic effect of CMCS was evaluated
using a classical apoptosis model induced by NO.
Chondrocyte apoptosis is key in the degeneration and
degradation of articular cartilage in cases of OA. Reduced
cellularity is a characteristic feature of OA cartilage, and
apoptosis has been suggested as an underlying cause (32).
In the present study, the results of a CCK-8 assay
revealed that cell viability in the articular chondrocytes
following SNP induction were markedly decreased, compared with
those in normal chondrocytes, and cell viability correlated
positively with the percentage of apoptotic chondrocytes. This
suggested that reduced cellularity in OA cartilage may, at least
partially, be attributed to cell death by apoptosis. The results
from the FCM showed that CMCS suppressed apoptosis of the
SNP-induced chondrocytes. These results were confirmed in the in
vitro experiments in the present study, which revealed that
CMCS inhibited the increase in the proportion of early and late
apoptotic cells in the NO-induced chondrocytes, suggesting that
CMCS prevented the degeneration and degradation of articular
cartilage by inhibiting chondrocyte apoptosis.
Apoptosis is an autonomous process of programmed
cell death, regulated by multiple signaling pathways. One of the
important pathways during the apoptotic process involves the
activation of caspase-3, and induction of the hydrolysis of nucleic
acids and cytoskeletal proteins (33). In vitro studies have
confirmed that caspase-3 activity is increased significantly in
NO-induced chondrocyte apoptosis (34,35).
Notably, caspase-3 is activated by cytochrome c (Cyt
c), which is induced by apoptotic signals. These signaling
events are set in motion by the pro-apoptotic protein, Bax, a
member of the Bcl-2 family, which migrates to the mitochondrial
membrane and induces the release of Cyt c (36,37).
Caspase-3 is a member of the downstream Cyt c signaling
pathway, and is the most important executor of cell apoptosis. A
previous study reported that millimeter wave treatment inhibited
p38/MAPK signaling and inhibited caspase-3 activation and
chondrocyte apoptosis (20).
Therefore, inhibiting the activation of caspase-3 may be the key to
decreasing chondrocyte apoptosis. The results of the present study
indicated that CMCS inhibited the activation of caspase-3 induced
by NO, and this may be one of the most important anti-apoptotic
mechanisms for CMCS.
The present study demonstrated that CMCS treatment
inhibited NO-induced effects on chondrocytes in vitro,
including apoptosis and the expression of caspase-3, and effects
involving inhibition of the p38/MAPK signaling pathway. CMCS (50,
100 and 200 µg/ml) protected chondrocytes from SNP-induced
apoptosis, and inhibited the apoptotic rates of NO-induced rat
chondrocytes, with effects on the expression levels of Bcl-2 and
Bax. CMCS also reduced caspase-3 activity in the SNP-induced
chondrocytes, and suppressed p38 phosphorylation, suggesting that
the p38/MAPK signaling pathway may represent a target for the
effects of CMCS, which may offer potential in the treatment of
OA.
There were several limitations to the present study.
Notably, all experiments were performed in a cell-based in
vitro system, and whether equivalent effects are be observed
consistently in an in vivo model and in patients remains to
be elucidated. Furthermore, all data were acquired with
CMCS-pretreated cells, which appeared to be essential for the
protective effects of the compound in the present study and in
previous reports (15–19). Further investigations, to examine
whether the pre-treated cells have altered signaling pathways in
SNP and other biological process, may assist in improving the
preparation of cells against apoptotic treatment.
In conclusion, the present study demonstrated that
CMCS inhibited SNP-induced chondrocyte apoptosis, promoted the
expression of Bcl-2, and inhibited the expression of Bax and
caspase-3 activity via its effects on the p38/MAPK signaling
pathway. Collectively, these results indicated that CMCS may have a
potential therapeutic role for the treatment of OA.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81301056) and the Natural
Science Foundation of Hubei Province (grant no. 2013CKB002).
References
|
1
|
Loeser RF: Aging and osteoarthritis: The
role of chondrocytes senescence and aging changes in the cartilage
matrix. Osteoarthritis Cartilage. 17:971–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thomas CM, Fuller CJ, Whittles CE and
Sharif M: Chondrocyte death by apoptosis is associated with the
initiation and severity of articular cartilage degradation. In J
Rheum Dis. 14:191–198. 2011. View Article : Google Scholar
|
|
3
|
Chen Q, Zhang B, Yi T and Xia C: Increased
apoptosis in human knee osteoarthritis cartilage related to the
expression of protein kinase B and protein kinase Cα in
chondrocytes. Folia Histochem Cytobiol. 50:137–143. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang JG, Xia C, Zheng XP, Yi TT, Wang XY,
Song G and Zhang B: 17β-Estradiol promotes cell proliferation in
rat osteoarthritis model chondrocytes via PI3K/Akt pathway. Cell
Mol Biol Lett. 16:564–575. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andjelkov N, Elvenes J, Knutsen G and
Johansen O: Beta-endorphin regulation of MAPKs in cultures human
articular chondrocytes: MAPK inhibitors prevent the increase of
IL-1 beta protein levels during beta-endorphin stimulation. Cell
Commun Adhes. 14:1–8. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding L, Guo D and Homandberg GA: The
cartilage chondrolytic mechanism of fibronectin fragments involves
MAP kanases: Comparison of three fragments and native fibronectin.
Osteoarthritis Cartilage. 16:1253–1262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kong D, Zheng T, Zhang M, Wang D, Du S, Li
X, Fang J and Cao X: Static mechanical stress induces apoptosis in
rat endplate chondrocytes through MAPK and mitochondria-dependent
caspase activation signaling pathways. PLoS One. 8:e694032013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Takebe K, Nishiyama T, Hayashi S,
Hashimoto S, Fujishiro T, Kanzaki N, Kawakita K, Iwasa K, Kuroda R
and Kurosaka M: Regulation of p38 MAPK phosphorylation inhibits
chondrocyte apoptosis in response to heat stress or mechanical
stress. Int J Mol Med. 27:329–335. 2011.
|
|
9
|
Wang H, Wang Z, Chen J and Wu J: Apoptosis
induced by NO via phosphorylation of p38 MAPK that stimulates
NF-kappa B, p53 and caspase-3 activation in rabbit articular
chondrocytes. Cell Biol Int. 31:1027–1035. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hamamura K, Goldring MB and Yokota H:
Involvement of p38 MAPK in regulation of MMP-13 mRNA in
chondrocytes in response to surviving stress to endoplasmic
reticulum. Arch Oral Biol. 54:279–286. 2009. View Article : Google Scholar :
|
|
11
|
Namdari S, Wei L, Moore D and Chen Q:
Reduced limb length and worsened osteoarthritis in adult mice after
genetic inhibition of p38 MAP kinase activity in cartilage.
Arthritis Rheum. 58:3520–3529. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sena P, Manfredini G, Benincasa M, Mariani
F, Smargiassi A, Catani F and Palumbo C: Up-regulation of the
chemo-attractive receptor ChemR23 and occurrence of apoptosis in
human chondrocytesisolated from fractured calcaneal osteochondral
fragments. J Anat. 224:659–668. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen Q, Gao Y, Kao X, Chen J, Xue W, Xiong
Y and Wang Z: SNP-induced apoptosis may be mediated with caspase
inhibitor by JNK signaling pathways in rabbit articular
chondrocytes. J Toxicol Sci. 37:157–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu SQ, Qiu B, Chen LY, Peng H and Du YM:
The effects of carboxymethylated chitosan on metalloproteinase-1,-3
and tissue inhibitor of metalloproteinase-1 gene expression in
cartilage of experimental osteoarthritis. Rheumatol Int. 26:52–57.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen Q, Liu SQ, Du YM, Peng H and Sun LP:
Carboxymethylchitosan protects rabbit chondrocytes from
interleukin-1beta-induced apoptosis. Eur J Pharmacol. 541:1–8.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He B, Liu SQ, Chen Q, Li HH, Ding WJ and
Deng M: Carboxymethylated chitosan stimulates proliferation of
Schwann cells in vitro via the activation of the ERK and Akt
signaling pathways. Eur J Pharmacol. 667:195–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tao HY, He B, Liu SQ, Wei AL, Tao FH, Tao
HL, Deng WX, Li HH and Chen Q: Effect of carboxymethylated chitosan
on the biosynthesis of NGF and activation of the Wnt/β-catenin
signaling pathway in the proliferation of Schwann cells. Eur J
Pharmacol. 702:85–92. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He B, Tao HY and Liu SQ: Neuroprotective
effects of carboxymethylated chitosan on hydrogen peroxide induced
apoptosis in Schwann cells. Eur J Pharmacol. 740:127–134. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He B, Tao H, Liu S and Wei A: Protective
effect of carboxymethylated chitosan on hydrogen peroxide-induced
apoptosis in nucleus pulposus cells. Mol Med Rep. 11:1629–1638.
2015.
|
|
20
|
Li X, Du M, Liu X, Wu M, Ye H, Lin J, Chen
W and Wu G: Millimeter wave treatment inhibits NO-induced apoptosis
of chondrocytes through the p38MAPK pathway. Int J Mol Med.
25:393–399. 2010.PubMed/NCBI
|
|
21
|
Liang Q, Wang XP and Chen TS: Resveratrol
protects rabbit articular chondrocyte against sodium
nitroprusside-induced apoptosis via scavenging ROS. Apoptosis.
19:1354–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cuenda A, Rouse J, Doza YN, Meier R, Cohen
P, Gallagher TF, Young PR and Lee JC: SB203580 is a specific
inhibitor of a MAP kinase homologue which is stimulated by cellular
stresses and interleukin-1. FEBS Lett. 364:229–233. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glynn RW, Miller N, Mahon S and Kerin MJ:
Expression levels of HER2/neu and those of collocated genes at
17q12–21, in breast cancer. Oncol Rep. 28:365–369. 2012.PubMed/NCBI
|
|
24
|
Kühn K and Lotz M: Mechanisms of sodium
nitroprusside-induced death in human chondrocytes. Rheumatol Int.
23:241–247. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Q, Mei X, Han G, Ling P, Guo B, Guo
Y, Shao H, Wang G, Cui Z, Bai Y and Xu F: Xanthan gum protects
rabbit articular chondrocytes against sodium nitroprusside-induced
apoptosis in vitro. Carbohydr Polym. 131:363–369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu X, Yang F, Song T, Zeng A, Wang Q, Sun
Z and Shen J: Synthesis of carboxymethylated and quaternized
chitosan and their therapeutic effect on nonalcoholic Fatty liver
disease. J Agric Food Chem. 59:10683–10692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nam JP, Park SC, Kim TH, Jang JY, Choi C,
Jang MK and Nah JW: Encapsulation of paclitaxel into lauric
acid-O-carboxymetlyl chitosan transferring micelles for hydrophobic
drug delivery and site-specific targeted delivery. Int J Pharm.
457:124–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blanco FJ, Ochs RL, Schwarz H and Lotz M:
Chondrocyte apoptosis induced by nitric oxide. Am J Pathol.
146:75–85. 1995.PubMed/NCBI
|
|
29
|
Lee HG and Yang JH: PCB126 induces
apoptosis of chondrocytes via ROS-dependent pathways.
Osteoarthritis Cartilage. 20:1179–1185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gungor I, Yilmaz A, Ozturk AM, Ergun MA,
Menevse S and Kaya K: Bupivacaine and levobupivacaine induce
apoptosis in rat chondrocyte cell cultures at ultra-low doses. Eur
J Orthop Surg Traumatol. 24:291–295. 2014. View Article : Google Scholar
|
|
31
|
Uehara Y, Hirose J, Yamabe S, Okamoto N,
Okada T, Oyadomari S and Mizuta H: Endoplasmic reticulum
stress-induced apoptosis contributes to articular cartilage
degeneration via C/EBP homologous protein. Osteoarthritis
Cartilage. 22:1007–1017. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hwang HS and Kim HA: Chondrocyte apoptosis
in the pathogenesis of osteoarthritis. Int J Mol Sci.
16:26035–26054. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J and Yuan J: Caspases in apoptosis and
beyond. Oncogene. 27:6194–6206. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang H, Wang Z, Chen J and Wu J: Apoptosis
induced by NO via phosphorylation of p38 MAPK that stimulates
NF-kappaB, p53 and caspase-3 activation in rabbit articular
chondrocytes. Cell Biol Int. 31:1027–1035. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sakata S, Hayashi S, Fujishiro T, Kawakita
K, Kanzaki N, Hashimoto S, Iwasa K, Chinzei N, Kihara S, Haneda M,
et al: Oxidative stress-induced apoptosis and matrix loss of
chondrocytes is inhibited by eicosapentaenoic acid. J Orthop Res.
33:359–365. 2015. View Article : Google Scholar
|
|
36
|
Antonsson B: Bax and other pro-apoptotic
Bcl-2 family 'killer-proteins' and their victim the mitochondrion.
Cell Tissue Res. 306:347–361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan TJ, Han LH, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar
|