Introduction
Colorectal cancer (CRC) is one of the most commonly
diagnosed malignant diseases worldwide, with greater than one
million new cases per year. Worldwide, it was the fourth leading
cause of cancer associated-mortality in 2011 (1,2). At
present, surgical resection is the primary treatment for localized
CRC. However, despite progress made in the early diagnosis and
treatment of CRC which has resulted in improved prognosis for
patients, the majority of patients that are not suitable for
surgery present with distant metastasis at the time of diagnosis
(3,4). Thus, the therapeutic options for
unresectable or metastatic CRC remain limited. Therefore, there is
an urgent requirement for novel agents for patients with CRC, in
order to achieve improved treatment outcomes.
Recently, natural products have gained extensive
attention as novel anti-cancer therapeutic agents for their
relatively few side effects (5).
Nitidine chloride (NC; Fig. 1A) is
a natural bioactive phytochemical alkaloid that was first derived
from the root of Zanthoxylum nitidum (Roxb). Previous
studies have demonstrated that NC has anti-oxidant, anti-fungal,
anti-inflammatory and analgesic functions (6,7). In
recent years, NC has been reported to possess anti-tumor activity
in various types of cancer. NC has been observed to induce
apoptosis and inhibit the proliferation and metastasis of renal
cancer (8,9). Additionally, NC was observed to
inhibit the proliferation of breast cancer and suppress its
migration and invasion via the c-Src/focal adhesion kinase
signaling pathway (10,11). In digestive system neoplasms,
studies have reported that NC inhibits the proliferation of
hepatocellular carcinoma (12),
and suppresses angiogenesis and the growth of gastric cancer via
the signal transducer and activator of transcription 3 signaling
pathway (13). However, no
evidence has been reported regarding the direct effect of NC on
apoptosis and proliferation in CRC and the mechanisms of this
effect.
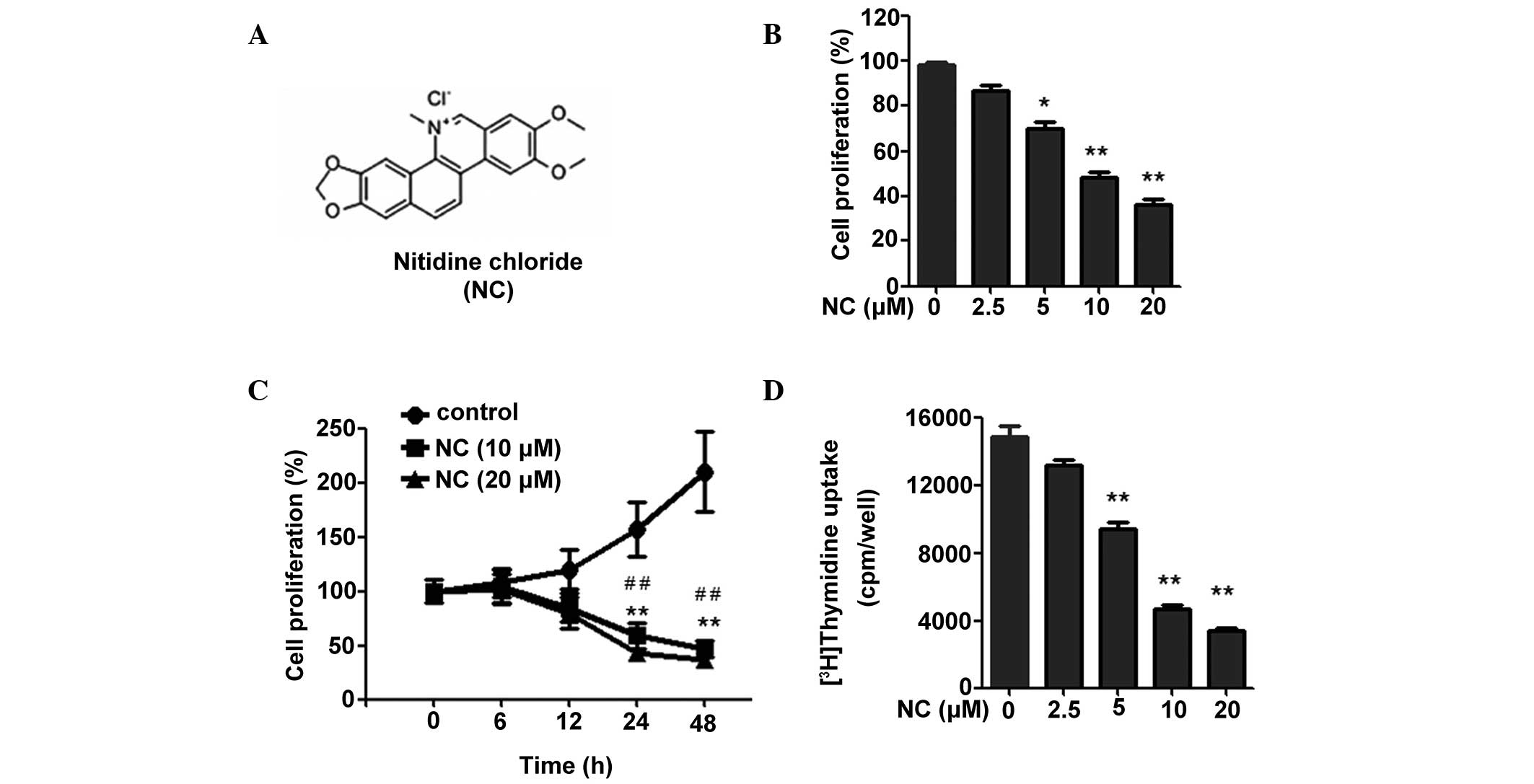 | Figure 1NC inhibits the proliferation of
HCT116 cells. (A) Chemical structure of NC. (B) Following treatment
with NC for 24 h (0, 2.5, 5, 10 and 20 µM), an MTT assay was
used to measure the proliferation of the HCT116 cells. (C)
Following treatment with 0, 10 and 20 µM NC for different
time durations (0, 6, 12, 24 and 48 h), an MTT assay was used to
detect the proliferation of the HCT116 cells. (D) Following
treatment with NC for 40 h at different concentrations (0, 2.5, 5,
10 and 20 µM), a 3H-thymidine uptake assay was
used to detect the proliferation of the HCT116 cells. The results
are presented as the mean ± standard deviation from three
independent experiments. *P<0.05,
**P<0.01 vs. the control group;
#P<0.05, ##P<0.01 vs. the cells treated
with 10 and 20 µM of NC. NC, nitidine chloride; MTT,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; cpm,
counts per minute. |
In the present study, the effects of NC on CRC cell
proliferation and apoptosis were evaluated, in addition to the
underlying molecular mechanisms.
Materials and methods
Cell lines and reagents
The HCT116 human CRC cell line was obtained from
American Type Culture Collection (Manassas, VA, USA) and cells were
routinely cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc, Waltham, MA, USA)
containing 10% fetal bovine serum (FBS; Invitrogen; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin, and 100 µg/ml
streptomycin (MACGENE Biotechnology, Ltd., Beijing, China) in 5%
CO2 at 37°C. The annexin V-fluorescein isothiocyanate
(FITC) apoptosis detection kit was purchased from BD Biosciences
(Franklin Lakes, NJ, USA). A terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) kit was purchased from Beyotime
Institute of Biotechnology (Shanghai, China). Monoclonal rabbit
anti-human extracellular signal-regulated kinase (ERK)1/2 (1:1,000;
cat. no. 4695), monoclonal rabbit anti-human phos-phorylated
(p)-ERK1/2 (1:1,000; cat. no. 4370), monoclonal rabbit anti-human
Bcl-2 (1:1,000; cat. no. 2870), monoclonal rabbit anti-human Bax
(1:1,000; cat. no. 5023), monoclonal rabbit anti-human caspase-9
(1:1,000; cat. no. 9502, monoclonal rabbit anti-human caspase-3
(1:1,000; cat. no. 9665) and monoclonal rabbit anti-human p53
antibodies (1:1,000; cat. no. 2527) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). NC was purchased
from Shanghai Tauto Biotech Co., Ltd. (Shanghai, China) and
dissolved in dimethyl sulfoxide (DMSO; Beijing Solarbio Science
& Technology Co., Ltd., Beijing, China). U0126, which is an
inhibitor of mitogen-activated protein kinase 1/2 located upstream
of ERK1 and ERK2 thus regulating ERK activity, was purchased from
Sigma-Aldrich (St. Louis, MO, USA).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay of cell viability and proliferation
Cell viability and proliferation were assessed by
MTT assay. HCT116 cells (5,000 cells/well) in 100 µl medium
were seeded into 96-well plates. Cells were pretreated with U0126
(10 µM) for 5 min. Following stimulation with NC of various
concentrations (0, 2.5, 5, 10 and 20 µM) for various
durations (0, 6, 12, 24 and 48 h), 20 µl MTT (5 mg/ml) was
added to each well. Following incubation for 4 h, 100 µl of
dimethyl sulfoxide (DMSO) was added to each well for a further 15
min. Finally, the absorbance values were determined using an
microplate luminometer (iMark; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) at 490 nm.
[3H] Thymidine uptake
HCT116 cells were cultured in DMEM with 10% FBS to
50% confluence. The cells were then cultured in serum-free DMEM for
24 h and treated with NC of various concentrations (0, 2.5, 5, 10,
20 µM) for 40 h. [3H] thymidine (final
concentration 1 uCi/ml; PerkinElmer, Inc., Waltham, MA, USA) was
added to the media during the last 24 h of culture. Following
washing with ice-cold phosphate-buffered saline (PBS), the cells
were precipitated with ice-cold 5% trichloroacetic acid (TCA;
Sigma-Aldrich) for a minimum of 4 h and washed twice with ice-cold
5% TCA followed by two washes with ice-cold PBS. Subsequently, the
cells were lysed with 200 µl 0.5 M NaOH for 30 min at 37°C.
DNA synthesis was measured by [3H] thymidine uptake
using a liquid scintillation counter (LS-6500; Beckman Coulter,
Inc., Brea, CA, USA).
Analysis of apoptosis by annexin
V-FITC/propidium iodide (PI) staining
Apoptosis in HCT116 cells was determined using an
annexin V-FITC/PI assay. In brief, HCT116 cells were seeded into
6-well plates at 1×106 cells/well. Following treatment
with NC (0, 10, 20 µM) for 24 h, HCT116 cells were then
harvested, washed and resuspended in PBS. Apoptotic cells were
determined with an annexin V-FITC apoptosis detection kit according
to the manufacturer's protocol. Briefly, the cells were washed and
subsequently incubated for 15 min at room temperature in the dark
in 200 µl 1X binding buffer containing 5 µl annexin
V-FITC and 10 µl PI. Apoptosis was determined by the BD
Accuri C6 Flow Cytometer (BD Biosciences) and processed using
Flowjo software (version 7.6.5; Flowjo LLC, Ashland, OR, USA).
Analysis of apoptosis by TUNEL
assays
The TUNEL method was used to label the apoptotic
cells using a TUNEL apoptosis detection kit (Beyotime Institute of
Biotechnology) according to the manufacturer's protocol. Briefly,
HCT116 cells were incubated with NC (0, 10, 20 µM) for 24 h.
Following incubation, the cells were fixed with 4% formaldehyde and
permeabilized with 0.1% Triton X-100 at 4°C for 10 min. The cells
were then stained by the TUNEL mixture for 1 h, followed by
staining with 4,6-diamidino-2-phenylindole (DAPI) for 5 min. The
TUNEL positive cells (red staining) were photographed and evaluated
qualitatively using Nikon Eclipse Ti (Nikon Corp, Tokyo, Japan) and
Ultra VIEW:emoji:VOX confocal microscopes (PerkinElmer, Inc.), and
images were analyzed by using Volocity software (version 6.0;
PerkinElmer, Inc.).
Western blot analysis
Protein was extracted from cells using a protein
lysis buffer (Beyotime Institute of Biotechnology) immediately
after the end of treatment. Total cell protein concentrations were
determined using a bicinchoninic acid protein assay kit (Pierce
Biotechnology, Inc., Rockford, IL, USA). Equal amounts of protein
(10 µg) from the cell lysates were run with 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (Bio-Rad
Laboratories, Inc.). Following electrophoresis, proteins were
transferred to polyvinylidene membranes (EMD Millipore, Billerica,
MA, USA), blocked with 5% fat-free milk at room temperature for 1
h, and incubated with the indicated primary antibodies overnight at
4°C. Subsequently, the membranes were washed with Tris-buffered
0.2% saline-Tween 20 and incubated with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibodies
(1:10,000; cat. no. ZDR-5306; Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) for 1 h at room
temperature. Immune complexes were detected with enhanced
chemiluminescence reagents (EMD Millipore, Boston, MA, USA), and
the blots were quantified by densitometric analysis using the Alpha
Imager 2200 (Genetic Technologies, Inc., Miami, FL, USA).
Statistical analysis
The data are expressed as the mean ± standard
deviation. All of the experiments were repeated a minimum of three
times. Comparisons between the group values were performed by
one-way analysis of variance. Holm's t-test was used for multiple
comparisons between the groups. All statistical analysis was
performed in SPSS (version 10.0; SPSS, Inc., Chicago, IL, USA)
P<0.05 was considered to indicate a statistically significant
difference.
Results
NC inhibits the proliferation of CRC
cells
To investigate the effect of NC on CRC cell
proliferation, an MTT assay was performed. As shown in Fig. 1B and C, NC inhibited the
proliferation of CRC cells in a time- and dose-dependent manner. To
further assess the result, the effects of NC on CRC cell
proliferation were detected by [3H] thymidine uptake.
The result demonstrated that NC is able to significantly inhibit
CRC cell proliferation over a range of concentrations (5, 10 and 20
µM; Fig. 1D). Considering
that the optimal inhibitory effect was observed when the NC
concentration was 10 and 20 µM, these concentrations of NC
were selected for the subsequent experiments. Taken together, these
data suggest that NC inhibits the proliferation of CRC cells.
NC induces apoptosis in CRC cells
To determine whether the NC-induced inhibition of
proliferation was due to a direct effect on apoptosis in CRC cells,
the cells were treated with 0, 10 and 20 µM NC for 24 h. As
indicated by the annexin V-FITC/PI assay, NC markedly increased
apoptosis in CRC cells at doses of 10 and 20 µM (Fig. 2A and B). The percentage of cells
undergoing apoptotic cell death increased from 6.9±1.3 to 34.8±6.8
and 36.9±7.2% following exposure to 10 and 20 µM NC,
respectively, for 24 h. TUNEL was used to detect the fragmented DNA
in cells undergoing apoptosis. Following NC treatment (0, 10 and 20
µM) for 24 h, the cells were stained with TUNEL and DAPI and
analyzed by fluorescence microscopy. As shown in Fig. 2C and D, 10 and 20 µM NC
increased the proportion of apoptotic cells. The significant
induction of apoptosis by NC treatment demonstrated its anti-cancer
effect on CRC cells.
 | Figure 2NC induced dose-dependent apoptosis in
HCT116 cells. (A) The HCT116 cells were treated with NC (0, 10 and
20 µM) for 24 h and then double-stained with PI and annexin
V. The cells were analyzed using a flow cytometer. (B) The bar
graph represents the results of three independent experiments. (C)
The HCT116 cells were treated with NC (0, 10 and 20 µM) for
24 h and then stained with DAPI and TUNEL, which labeled the 3′-OH
ends of the fragmented DNA in apoptotic cells, and were viewed
using a confocal microscope (magnification, x200). (D)
Quantification of the percentage of TUNEL positive cells. The bar
graph represents the results of three independent experiments.
**P<0.01 vs. the control group. NC, nitidine
chloride; PI, propidium iodide; DAPI,
4′,6-diamidino-2-phenylindole; TUNEL, terminal deoxynucleotidyl
transferase dUTP nick end labeling. |
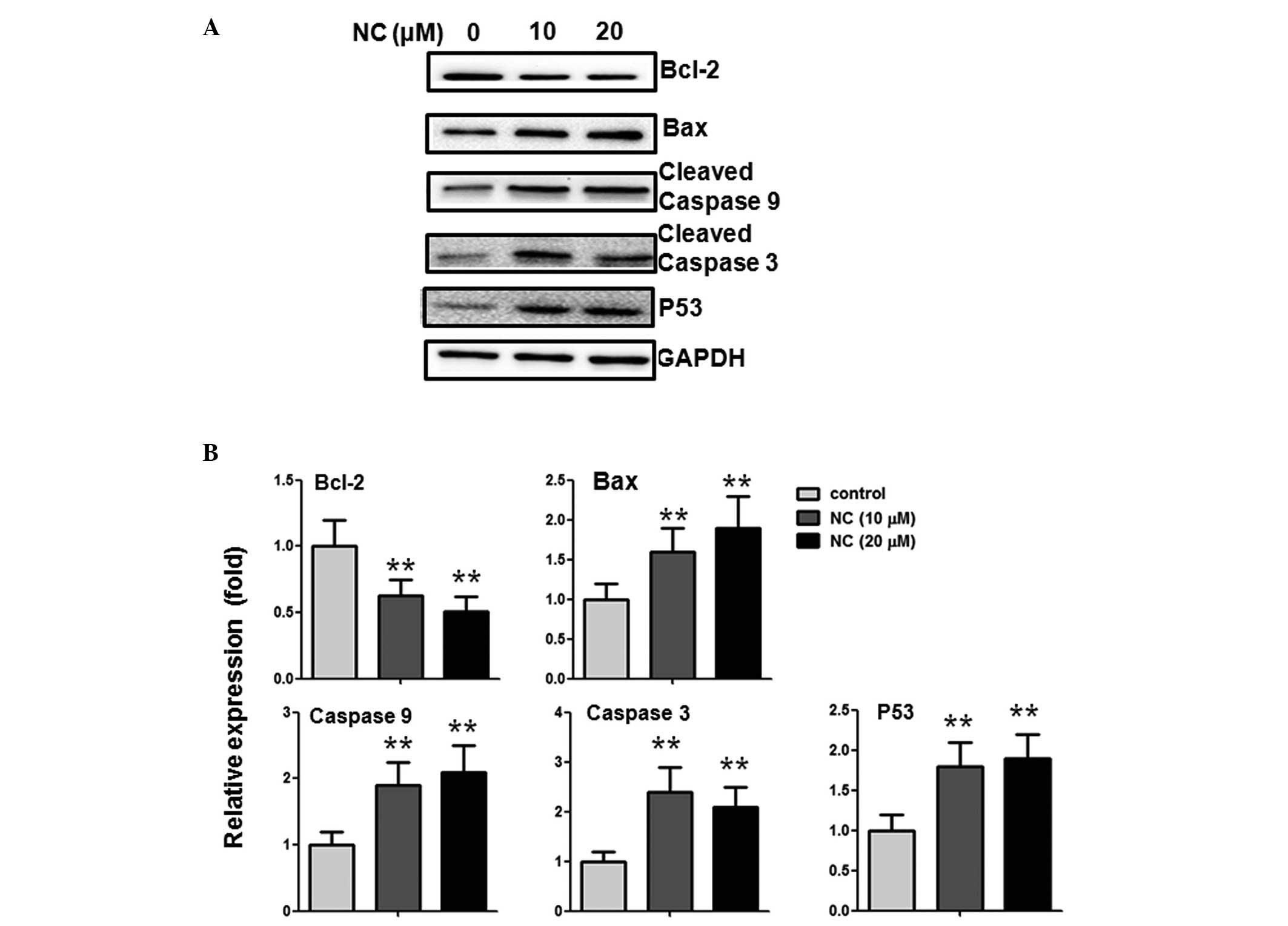
NC induces apoptosis in CRC cells via
alterations of Bcl-2 family proteins and caspase activation
To further investigate the potential mechanism of
NC-induced CRC cell apoptosis, the impact of NC on the expression
levels of Bcl-2, Bax, p53, caspase-3 and -9 were examined. The
western blotting results indicated that, following treatment with
10 and 20 µM NC, the expression of the anti-apoptotic
protein, Bcl-2, was reduced and the pro-apoptotic protein Bax was
increased. In addition, the expression levels of cleaved caspase-3
and -9, and p53 were upregulated (Fig.
3A and B).
NC inhibits ERK phosphorylation in CRC
cells
The ERK pathway serves an important role in tumor
development. The inhibition of ERK pathway activation has been
demonstrated to induce tumor cell apoptosis (14). To gain further insight into the
association between NC and the proliferation and apoptosis of CRC
cells, ERK signaling molecules were investigated. As presented in
Fig. 4A, ERK phosphorylation was
reduced following treatment with 10 and 20 µM NC for 24 h,
indicating that the ERK pathway may be involved in CRC cell
apoptosis. The reduction was statistically significant, as
quantified by densitometry (Fig.
4B).
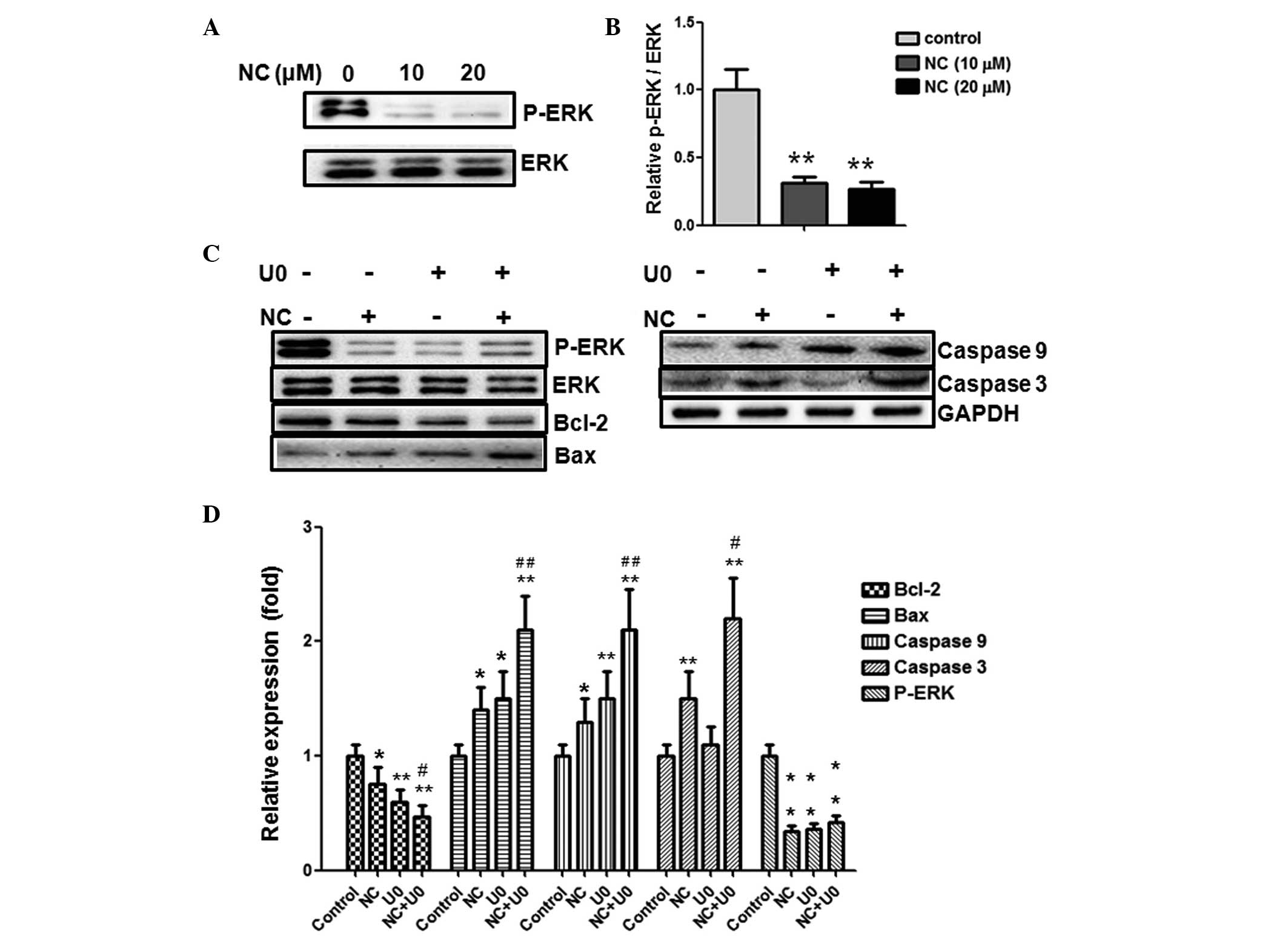 | Figure 4Pro-apoptotic effect of NC on HCT116
cells is ERK-dependent. (A) The HCT116 cells were treated with NC
(0, 10 and 20 µM) for 24 h, and the levels of p-ERK1/2,
ERK1/2 were analyzed by western blotting. (B) Quantification of the
western blotting results. (C) The HCT116 cells were treated with NC
(20 µM) and/or U0126 (10 µM) for 24 h, the levels of
p-ERK1/2, ERK1/2, Bcl-2, Bax, cleaved caspase-3 and -9 were
analyzed by western blotting with GAPDH as a control. (D)
Quantification of the western blotting results. Data are presented
as the mean ± standard deviation from three independent
experiments.*P<0.05, **P<0.01 vs. the
control group. #P<0.05, ##P<0.01 vs.
the cells treated with NC. NC, nitidine chloride; ERK,
extracellular-signal-regulated kinase; p, phosphorylated; GAPDH,
glyceraldehyde 3-phosphate dehydrogenase; U0, U0126. |
Inhibition of the ERK pathway enhances
the pro-apoptotic and anti-proliferative effects of NC in CRC
cells
To further investigate whether the effects of NC are
ERK-dependent, U0126 (MEK1/2 inhibitor which are upstream of ERK,
therefore results in ERK inhibition) was used to prevent ERK
activation. As shown in Fig. 4C and
D, inhibition of ERK activity using U0126 enhanced the
upregulation of Bax, cleaved caspase-3 and -9 expression and the
downregulation of Bcl-2 expression induced by NC. This demonstrated
that NC-induced apoptosis may be ERK dependent. To further
investigate the anti-proliferative effects of NC, the proliferation
of CRC cells was measured using an MTT assay. As shown in Fig. 5A, the ERK inhibitor significantly
enhanced the NC-induced inhibition of CRC cell proliferation.
Similar results were observed using [3H] thymidine
uptake (Fig. 5B), which
demonstrated that preventing ERK activation was able to
significantly enhance the NC-induced inhibition of CRC cell
proliferation.
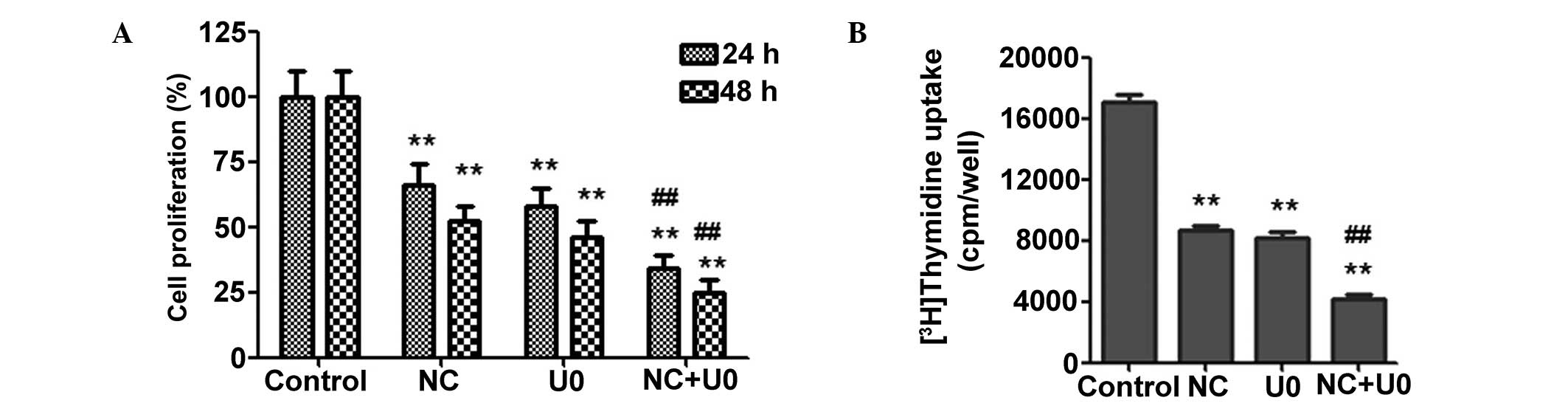 | Figure 5NC inhibition of HCT116 cell
proliferation is ERK-dependent. (A) The HCT116 cells were treated
with NC (20 µM) and/or U0126 (10 µM) for 24 h and 48
h, and cell viability was measured by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay.
(B) The HCT116 cells were treated with NC (20 µM) and/or
U0126 (10 µM) for 40 h, and cell proliferation was measured
by 3H-thymidine uptake assay. The results are presented
as the mean ± standard deviation from three independent
experiments. *P<0.05, **P<0.01 vs. the
control group; #P<0.05, ##P<0.01 vs.
the cells treated with NC. NC, nitidine chloride; ERK,
extracellular-signal-regulated kinase; U0, U0126; cpm, counts per
minute. |
Discussion
Uncontrolled proliferation and/or resistance to
apoptosis, provides cancer cells with a survival advantage to
resist conventional chemotherapeutic agents (15,16).
In addition, conventional chemotherapeutic drugs are limited for
long-term use due to their toxicity and side-effects on normal
cells. In recent years, increasing attention has been focused on
the therapeutic functions of natural products in cancer therapy,
which have fewer side-effects (17–19).
As a natural, bioactive phytochemical alkaloid extracted from
Zanthoxylum nitidum (Roxb), NC has exhibited a wide range of
pharmacological activity against inflammation and oxidation
(6,7). In recent years, increasing evidence
suggests that NC has the ability to induce apoptosis and/or inhibit
the proliferation, migration and invasion of renal, breast,
hepatocellular and gastric cancer cells (8–13).
Therefore, NC is a promising chemotherapeutic agent for various
types of cancer. However, although previous studies have shed light
on the antitumor activity of NC, whether NC has any effect on the
proliferation and apoptosis of CRC cells remains unknown, as do the
detailed molecular mechanisms involved. The current study, to the
best of our knowledge, was the first demonstration that NC is able
to inhibit proliferation and induce apoptosis in CRC cells. In
addition, NC was demonstrated to have this effect on CRC cells via
the ERK signaling pathway. These data provide a novel molecular
mechanism by which NC exerts its anti-cancer effect in CRC
cells.
Apoptosis is regulated by pro-apoptotic and
anti-apoptotic proteins, and is executed through caspases (20). The balance of Bcl-2 protein family
members, including anti-apoptotic proteins (Bcl-2, Bcl-xL) and
pro-apoptotic proteins (Bax, Bad), serves a crucial role in
modulating and executing a number of apoptotic pathways (21,22).
In previous studies, the anti-apoptotic Bcl-2 protein has been
shown to function as a preserver of the mitochondrial membrane,
preventing the release of internal calcium into the cytoplasm and
inhibiting the oligomerization of the anti-apoptotic Bax protein
(23,24). Bax has been shown to translocate
into mitochondria to regulate cytochrome C release, thus resulting
in the activation of caspase-3 and -9, and inducing apoptosis
(25,26).
As reported in previous studies (8,11),
NC exhibits potent activity in inhibiting the proliferation of
cancer cells by inducing apoptosis and modulating the expression of
the Bcl-2 family. In the present study, NC was observed to induce
apoptosis and inhibit proliferation in CRC cells, in a dose- and
time-dependent manner. Furthermore, the expression of the
pro-apoptotic protein, Bax, and the anti-apoptotic protein, Bcl-2,
was measured in CRC cells treated with NC. NC was observed to
upregulate the expression of Bax and downregulate the expression of
Bcl-2. Additionally, caspase-3 and -9 were further activated by NC
treatment, resulting in apoptosis. These results suggest that
NC-induced CRC cell apoptosis may be attributed to the reduced
expression of Bcl-2 and the increased expression of Bax, resulting
in the activation of caspase-3 and -9. A previous study
demonstrated that the role of the tumor suppressor p53 in apoptosis
was associated with several Bcl-2 family members (27). In the present study, p53 expression
in CRC cells was measured following treatment with NC, which
indicated that NC upregulated the expression of p53 in a
dose-dependent manner. This result further demonstrates that p53
was activated in the NC-induced apoptosis of CRC cells.
As a key signal transduction pathway, the ERK
signaling pathway is involved in the regulation of proliferation,
differentiation, senescence and apoptosis in cancer cells (28). Previous studies have revealed that
cell survival and apoptosis are modulated by the ERK signaling
pathway (29,30). In the present study, the ERK
activity in CRC cells was detected by western blot analysis. The
results showed that the expression of p-ERK was reduced following
treatment with NC. To confirm that the ERK signaling pathway was
involved in the NC-induced apoptosis and the inhibition of
proliferation in CRC cells, ERK phosphorylation was prevented using
U0126, the inhibitor of MEK1/2, which are upstream of ERK,
therefore resulting in ERK inhibition.. This indicated that the
inhibition of ERK activity by U0126 enhanced the upregulation of
Bax, caspase-3 and -9, and the downregulation of Bcl-2 induced by
NC. In addition, ERK inhibition resulted in the inhibition of cell
proliferation. These results suggest that NC induced the apoptosis
of CRC cells through the suppression of ERK activity, and by
altering the expression of Bax and Bcl-2. Furthermore, ERK
inhibition by U0126 enhanced the increased expression of Bax and
reduced expression of Bcl-2 induced by NC, further indicating that
ERK was upstream of Bax and Bcl-2.
In conclusion, the present study demonstrated that
NC inhibits the proliferation of CRC cells and induces apop-tosis.
Furthermore, the effect of NC was mediated through the ERK
signaling pathway. Therefore, the current study suggests that NC is
a potential therapeutic agent for the treatment of colorectal
cancer. Further in vivo studies should be performed to
confirm these results and investigate the effects further.
Acknowledgments
The current study was supported by the Natural
Science Foundation of China (grant no. 81370325) and the Yantai
Scientific Development Project (grant no. 2013WS216).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Center MM, Jemal A, Smith RA and Ward E:
Worldwide variations in colorectal cancer. CA Cancer J Clin.
59:366–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cunningham D, Atkin W, Lenz HJ, Lynch HT,
Minsky B, Nordlinger B and Starling N: Colorectal cancer. Lancet.
375:1030–1047. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang WQ, Fu FF, Li YX, Wang WB, Wang HH,
Jiang HP and Teng LS: Molecular biomarkers of colorectal cancer:
Prognostic and predictive tools for clinical practice. J Zhejiang
Univ Sci B. 13:663–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Jiang W, Zhang Z, Qian M and Du B:
Nitidine chloride inhibits LPS-induced inflammatory cytokines
production via MAPK and NF-kappaB pathway in raw 264.7 cells. J
Ethnopharmacol. 144:145–150. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Del Poeta M, Chen SF, Von Hoff D, Dykstra
CC, Wani MC, Manikumar G, Heitman J, Wall ME and Perfect JR:
Comparison of in vitro activities of camptothecin and nitidine
derivatives against fungal and cancer cells. Antimicrob Agents
Chemother. 43:2862–2868. 1999.PubMed/NCBI
|
|
8
|
Fang Z, Tang Y, Jiao W, Xing Z, Guo Z,
Wang W, Xu Z and Liu Z: Nitidine chloride induces apoptosis and
inhibits tumor cell proliferation via suppressing ERK signaling
pathway in renal cancer. Food Chem Toxicol. 66:210–216. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fang Z, Tang Y, Jiao W, Xing Z, Guo Z,
Wang W, Shi B, Xu Z and Liu Z: Nitidine chloride inhibits renal
cancer cell metastasis via suppressing AKT signaling pathway. Food
Chem Toxicol. 60:246–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pan X, Han H, Wang L, Yang L, Li R, Li Z,
Liu J, Zhao Q, Qian M, Liu M and Du B: Nitidine chloride inhibits
breast cancer cells migration and invasion by suppressing c-Src/FAK
associated signaling pathway. Cancer Lett. 313:181–191. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun M, Zhang N, Wang X, Cai C, Cun J, Li
Y, Lv S and Yang Q: Nitidine chloride induces apoptosis, cell cycle
arrest, and synergistic cytotoxicity with doxorubicin in breast
cancer cells. Tumour Biol. 35:10201–10212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao J, Xu T, Zheng JX, Lin JM, Cai QY, Yu
DB and Peng J: Nitidine chloride inhibits hepatocellular carcinoma
cell growth in vivo through the suppression of the JAK1/STAT3
signaling pathway. Int J Mol Med. 32:79–84. 2013.PubMed/NCBI
|
|
13
|
Chen J, Wang J, Lin L, He L, Wu Y, Zhang
L, Yi Z, Chen Y, Pang X and Liu M: Inhibition of STAT3 signaling
pathway by nitidine chloride suppressed the angiogenesis and growth
of human gastric cancer. Mol Cancer Ther. 11:277–287. 2012.
View Article : Google Scholar
|
|
14
|
Sebolt-Leopold JS and English JM:
Mechanisms of drug inhibition of signalling molecules. Nature.
441:457–462. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cai Q, Lin J, Wei L, Zhang L, Wang L, Zhan
Y, Zeng J, Xu W, Shen A, Hong Z and Peng J: Hedyotis diffusa Willd
inhibits colorectal cancer growth in vivo via inhibition of STAT3
signaling pathway. Int J Mol Sci. 13:6117–6128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan AC, Konczak I, Sze DM and Ramzan I:
Molecular pathways for cancer chemoprevention by dietary
phytochemicals. Nutr Cancer. 63:495–505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thomasset SC, Berry DP, Garcea G, Marczylo
T, Steward WP and Gescher AJ: Dietary polyphenolic
phytochemicals-promising cancer chemopreventive agents in humans? A
review of their clinical properties. Int J Cancer. 120:451–458.
2007. View Article : Google Scholar
|
|
20
|
Zhang Y, Zhuang Z, Meng Q, Jiao Y, Xu J
and Fan S: Polydatin inhibits growth of lung cancer cells by
inducing apoptosis and causing cell cycle arrest. Oncology Lett.
7:295–301. 2014.
|
|
21
|
Reed JC: Bcl-2: Prevention of apoptosis as
a mechanism of drug resistance. Hematol Oncol Clin North Am.
9:451–473. 1995.PubMed/NCBI
|
|
22
|
Brady HJ and Gil-Gómez G: Bax. The
pro-apoptotic Bcl-2 family member, Bax. Int J Biochem Cell Biol.
30:647–650. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baffy G, Miyashita T, Williamson JR and
Reed JC: Apoptosis induced by withdrawal of interleukin-3 (IL-3)
from an IL-3-dependent hematopoietic cell line is associated with
repartitioning of intracellular calcium and is blocked by enforced
Bcl-2 oncoprotein production. J Biol Chem. 268:6511–6519.
1993.PubMed/NCBI
|
|
24
|
Precht TA, Phelps RA, Linseman DA, Butts
BD, Le SS, Laessig TA, Bouchard RJ and Heidenreich KA: The
permeability transition pore triggers Bax translocation to
mitochondria during neuronal apoptosis. Cell Death Differ.
12:255–265. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Antonsson B: Bax and other pro-apoptotic
Bcl-2 family 'killer-proteins' and their victim the mitochondrion.
Cell Tissue Res. 306:347–361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Crompton M: Bax, Bid and the
permeabilization of the mitochondrial outer membrane in apoptosis.
Curr Opin Cell Biol. 12:414–419. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu Q: Restoring p53-mediated apoptosis in
cancer cells: New opportunities for cancer therapy. Drug Resist
Updat. 9:19–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Thompson N and Lyons J: Recent progress in
targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug
discovery. Curr Opin Pharmacol. 5:350–356. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gendron S, Couture J and Aoudjit F:
Integrin alpha2beta1 inhibits Fas-mediated apoptosis in T
lymphocytes by protein phosphatase 2A-dependent activation of the
MAPK/ERK pathway. J Biol Chem. 278:48633–48643. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shelton JG, Steelman LS, White ER and
McCubrey JA: Synergy between PI3K/Akt and Raf/MEK/ERK pathways in
IGF-1R mediated cell cycle progression and prevention of apoptosis
in hematopoietic cells. Cell Cycle. 3:372–379. 2004.PubMed/NCBI
|