Introduction
Cardiopulmonary bypass (CPB) has been widely used in
open heart surgery in the last six decades since John H. Gibbon
invented the artificial heart and lung machine (1). With improvements in medical equipment
and biomaterial technologies, including smaller prime volume
circuits, more biocompatible surfaces and gas-permeable microporous
membranes, the incidence of CPB-induced complications have
significantly decreased (2,3).
However, CPB is known to activate systemic inflammatory response
syndrome with acute lung injury that is associated with
microvascular barrier injury (4).
Numerous factors, including pulmonary hypoperfusion, induction of
inflammatory mediators, hypothermia and blood contact with foreign
surfaces during CPB, contribute to the etiology of lung injury
(5). Post-surgical lung injury
predominantly consists of lung edema and hypoxia, which are
associated with CPB-induced neutrophil infiltration and increased
microvascular permeability (6).
The Src family is important in intracellular signal
transduction in acute inflammatory responses (7,8). Src
is widely expressed by macrophages, monocytes, neutrophils,
alveolar epithelial cells, endothelial cells and fibroblasts in the
lung. It has been reported that Src is involved in the increase of
lung vascular permeability in mice exposed to mechanical
ventilation and hyperoxia-augmented ventilation (9,10).
Thus, the present study aimed to determine whether the Src kinase
pathway is involved in CPB-induced proinflammatory cytokine
secretion, neutrophil infiltration and microvascular
hyperpermeability.
Caveolin-1, a member of the caveolin family, exists
primarily in lung endothelial cells and type I epithelial cells and
functions as a structural and signaling protein (11). It is required for the formation and
trafficking of caveolae, the primary vesicular carriers and
mechanism of transcellular macromolecule transport through the
vascular endothelial barrier (12,13).
Vascular endothelial cadherin (VE-cadherin) is a cell-specific
member of the cadherin protein family, which regulates endothelial
adherens junctions (14,15). The present study initially
investigated the effects of CPB on pulmonary microvascular
permeability, neutrophil infiltration and secretion of
proinflammatory cytokines. Subsequently, the role of Src kinase
activation, caveolin-1 and VE-cadherin phosphorylation in CPB was
examined.
Materials and methods
Animals and drugs
A total of 460 male Sprague-Dawley rats, weighing
250±10 g, age 10 weeks, were obtained from the Shanghai
Experimental Animal Center (Shanghai, China) and used in all
experiments. Animals were raised under standard conditions (22°C,
33% humidity) with a 12 h light/dark cycle. The study was performed
in accordance with the National Institute of Health Guide for the
Care and Use of Laboratory Animals (16) and with the approval of the research
committee at Shanghai Jiaotong University (Shanghai, China). The
non-specific Src kinase inhibitor 4-amino-5-(4-chlorophen
yl)-7-(dimethylethyl)pyrazolo[3,4-d]pyrimidine (PP2) was purchased
from Cayman Chemical Co. (Ann Arbor, MI, USA).
Experimental design
In order to examine the time course of pulmonary
microvascular permeability, neutrophil infiltration and
proinflammatory cytokine secretion, rats were randomly assigned to
the following four groups (n=10 in each group): Sham group, CPB
group, CPB + PP2 group and the untreated group. Rats in the sham
group received similar surgery to the CPB and CPB + PP2 group,
however, no blood was drained from the rats. Rats in the CPB group
received CPB surgery as described below. Rats in the CPB + PP2
group received PP2 administration (1 mg/kg, intraperitoneal
injection) 30 min prior to CPB surgery. Rats in the untreated group
received no treatment. In order to investigate the time course of
Src kinase phosphorylation, rats were randomly assigned to five
groups (n=10 in each group): Pre-CPB (baseline), 0 h after CPB, 12
h after CPB, 24 h after CPB and 48 h after CPB. To determine the
alterations in VE-cadherin and caveolin-1 phosphorylation, rats
were randomly assigned to five groups (n=10 in each group): Sham,
24 h after CPB, 24 h after CPB + 1 mg/kg PP2, 24 h after CPB + 2
mg/kg PP2 and 24 h after CPB + 4 mg/kg PP2.
CPB procedure
The CPB procedure was performed according to the
method described in our previous study (17). Initially, animals were anesthetized
by intraperitoneal administration of butylone (60 mg/kg; Shanghai
Experimental Animal Center) and then pentobarbital (3%; 1.5 mg/kg
body weight; Shanghai Experimental Animal Center) was continuously
provided to maintain anesthesia. The right femoral artery was
cannulated with a 24-gauge catheter (heparinized with
polytetrafluoroethylene) to monitor arterial pressure. Following
administration of heparin (250 U/kg), a 16-gauge catheter was
advanced to the right atrium through the right jugular vein. A
22-gauge catheter was cannulated to the tail artery as an arterial
infusion line.
As described in our previous study (17), the mini-CPB circuit consisted of a
venous reservoir, a specially designed membrane oxygenator, a
roller pump and sterile polyvinyl chloride tubing with an internal
diameter of 3 mm for the venous and arterial lines. The roller pump
was equipped with a silicone tube 15 cm in length with an internal
diameter of 5 mm. The membrane oxygenator was specially designed
with a surface area for gas exchange of 0.05 m2
(Micro-1; Dongguan Kewei Medical Instrument Co., Ltd., Dongguan,
China), with a total assembly dynamic priming volume of ~2 ml. The
CPB circuit was primed with 12 ml of a solution of heparin (250
U/kg) and hetastarch. The blood was drained from the right atrium
through the jugular vein catheter to a 5-ml sterile open reservoir
using a siphon. A roller pump (BT00-300M; Baoding Lange Co., Ltd.,
Baoding, China) was used to drive the blood through silicone
arterial inflow tubing and then return it to the tail artery.
Evans blue dye (EBD) exclusion
analysis
Pulmonary microvascular injury was assessed by the
extravasation of EBD into the lung parenchyma as described by
Cavriani et al (18). EBD
solution (100 mg/ml) was prepared in phosphate-buffered saline
(PBS; pH 7.4) and intravenously injected at a dose of 30 mg/kg,
then allowed to circulate for 30 min prior to sacrifice by
decapitation. The right lungs were then excised and flushed with
cold PBS three times. Two samples of lung parenchyma were resected
and weighed. One sample was dried in an oven (60°C) for 72 h to
obtain the dry weight. The other sample was homogenized in 5 ml of
formamide to extract EBD. This homogenate was then incubated at
60°C for 24 h and centrifuged at 4,000 × g for 30 min. The
supernatant was then collected. The EBD optical density was
measured at a wavelength of 620 nm using an EAR 340 mictrotiter
plate reader (SLT-Lab Instruments, Salzburg, Austria). The
concentration of EBD was calculated from a standard curve of
EBD-formamide solutions. The dry/wet ratio of each lung sample was
calculated and used in the final calculation of Evans blue
extravasation. EBD was expressed as µg Evans blue/g dry
weight.
Bronchoalveolar lavage fluid (BALF)
collection and assays
At the time point of sample collection, animals were
sacrificed by decapitation and the chest was opened. A cannula was
then inserted into the left trachea. The left lung cavity was
gently flushed with 500 µl saline (4°C) up to a total volume
of 2 ml to obtain BALF, which was then centrifuged at 400 × g for
10 min. The supernatant was used for the proinflammatory cytokine
assay. The pelleted cells were re-suspended in PBS and then the
neutrophil count was determined using a Hemovet HV950FS (CDC
Technologies Inc., Oxford, CT, USA). ELISA kits (BioLegend, Inc.,
San Diego, CA, USA) were used to measure the levels of tumor
necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 in the
supernatants of BALF according to the manufacturer's protocol. The
results were expressed as pg/ml BALF.
Western blot analysis
Following collection of BALF, the lung tissues were
washed in ice-cold saline, then homogenized in 4°C RIPA lysis
buffer (Beyotime Institute of Biotechnology, Shanghai, China) with
1 mM phenylmethanesulfonyl fluoride and centrifuged at 3,000 × g
and 4°C for 15 min. The supernatants were collected and the protein
concentration was determined using a BCA protein assay kit
(Beyotime Institute of Biotechnology). Protein samples (40
µg) were loaded per lane and separated using 10% SDS-PAGE
(Beyotime Institute of Biotechnology). The target proteins,
including phosphorylated Src, VE-cadherin and caveolin-1, were then
electrophoretically transferred onto nitrocellulose membranes
(Beyotime Institute of Biotechnology). The protein blots were
blocked in Tris-Buffered Saline and Tween 20 (TBST; 5% non-fat
milk, 10 mM Tris, 150 mM NaCl, 0.05% Tweek-20) for 1 h, followed by
incubation with primary antibodies against phosphorylated Src
(monoclonal; 1:200, rabbit anti-mouse; ab4816; Oncogene Research
Products; La Jolla, CA, USA), phosphorylated VE-cadherin
(polyclonal, 1:400; rabbit anti-mouse; SAB4504676; BD Biosciences,
Franklin Lakes, NJ, USA), VE-cadherin (monoclonal; 1:400; rabbit
anti-mouse; V1514; BD Biosciences), phosphorylated caveolin-1
(polyclonal, 1:400; rabbit anti-mouse; sc-14037; Santa Cruz
Biotechnology Inc., Dallas, TX, USA) or caveolin-1 (monoclonal;
1:400; rabbit anti-mouse; sc-53564; Santa Cruz Biotechnology Inc.)
overnight at 4°C. Blots were then treated with the following
secondary antibodies in TBST solution for 1 h: Secondary Src
antibody [polyclonal; 1:4000; chicken anti-rabbit immunoglobulin G
(IgG); ab6829; Abcam, Cambridge, MA, USA], phosphorylated
VE-cadherin (polyclonal), VE-cadherin (monoclonal), phosphorylated
caveolin-1 (polyclonal) or caveolin-1 (monoclonal; all 1:3,000;
chicken anti-rat; ab112448; Abcam). Each sample was also probed
with β-actin antibody (1:30,000; rabbit anti-mouse; A5316l;
Sigma-Aldrich, St. Louis, MO, USA) as a loading control, and
β-actin secondary antibody (monoclonal; 1:3,000; chicken anti-mouse
IgG; ab131368, Abcam). Finally, blots were washed with PBS with
Tween 20 and then examined using the ECL Plus Western Blotting
Detection System (Amersham Life Science, Little Chalfont,
Buckinghamshire, UK).
Statistical analysis
Values are presented as the mean ± standard error of
the mean. Statistical analysis was performed using SPSS 17.0 (SPSS,
Inc., Chicago, IL, USA) with one-way analysis of variance followed
by Student-Newman-Keuls post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
PP2 attenuates the increase in pulmonary
microvascular leakage in BALF following CPB
Fig. 1 shows
alterations in pulmonary microvascular leakage over time
demonstrated by the concentration of EBD. Pulmonary microvascular
leakage increased up to 24 h after CPB, but decreased at 48 h after
CPB. Treatment with PP2 significantly inhibited the increase in
pulmonary microvascular leakage compared with the CPB group
(P<0.05). However, the CPB + PP2 group exhibited increased
pulmonary microvascular leakage (P<0.05) compared with the sham
group with the exception of at 0 h after CPB.
 | Figure 1EBD concentration in BALF following
CPB. Animals were sacrificed at different time points prior to or
following CPB surgery (Pre-CPB, 0 h after CPB, 12 h after CPB, 24 h
after CPB and 48 h after CPB). In the CPB + PP2 group, 1 mg/kg PP2
was intraperitoneally injected 30 min prior to CPB.
&P<0.05, compared with the sham group;
#P<0.05, compared with the CPB + PP2 group. EBD,
Evans Blue Dye; BALF, bronchoalveolar lavage fluid; CPB,
cardiopulmonary bypass; PP2, 4-amino-5-(4-chlorophenyl)-7-(di
methylethyl)pyrazolo[3,4-d]pyrimidine. |
PP2 attenuates increases in the
neutrophil count and proinflammatory cytokines, IL-1β and IL-6, in
BALF following CPB
Fig. 2A shows
alterations in the neutrophil count in BALF following CPB. The
neutrophil count was increased following CPB surgery over time
until 12 h after CPB. It peaked at 12 and 24 h after CPB and then
decreased at 48 h after CPB. Treatment with PP2 significantly
inhibited the increase in neutrophil count in BALF compared with
the CPB group (P<0.05, compared with the CPB group; P>0.05,
compared to the sham group). No significant difference was observed
between the sham group and the untreated group.
Fig. 2B–D show the
results of TNF-α, IL-1β and IL-6 in BALF. TNF-α partially increased
following CPB surgery, continued increasing in the first 24 h and
then decreased at 48 h after CPB. Treatment with PP2 did not alter
post-surgical increases in TNF-α. Unlike TNF-α, IL-1β increased
over time until 48 h after CPB. PP2 significantly inhibited the
level of IL-1β compared with the CPB group (P<0.05), although it
remained higher than the sham group (P<0.05). Alterations in
IL-6 concentration demonstrated a similar pattern with TNF-α and
peaked at 24 h after CPB. PP2 significantly ameliorated the
increase in IL-6 in BALF (P<0.05, compared with the CPB group;
P>0.05 compared with the sham group). No significant difference
was identified between the sham group and untreated group.
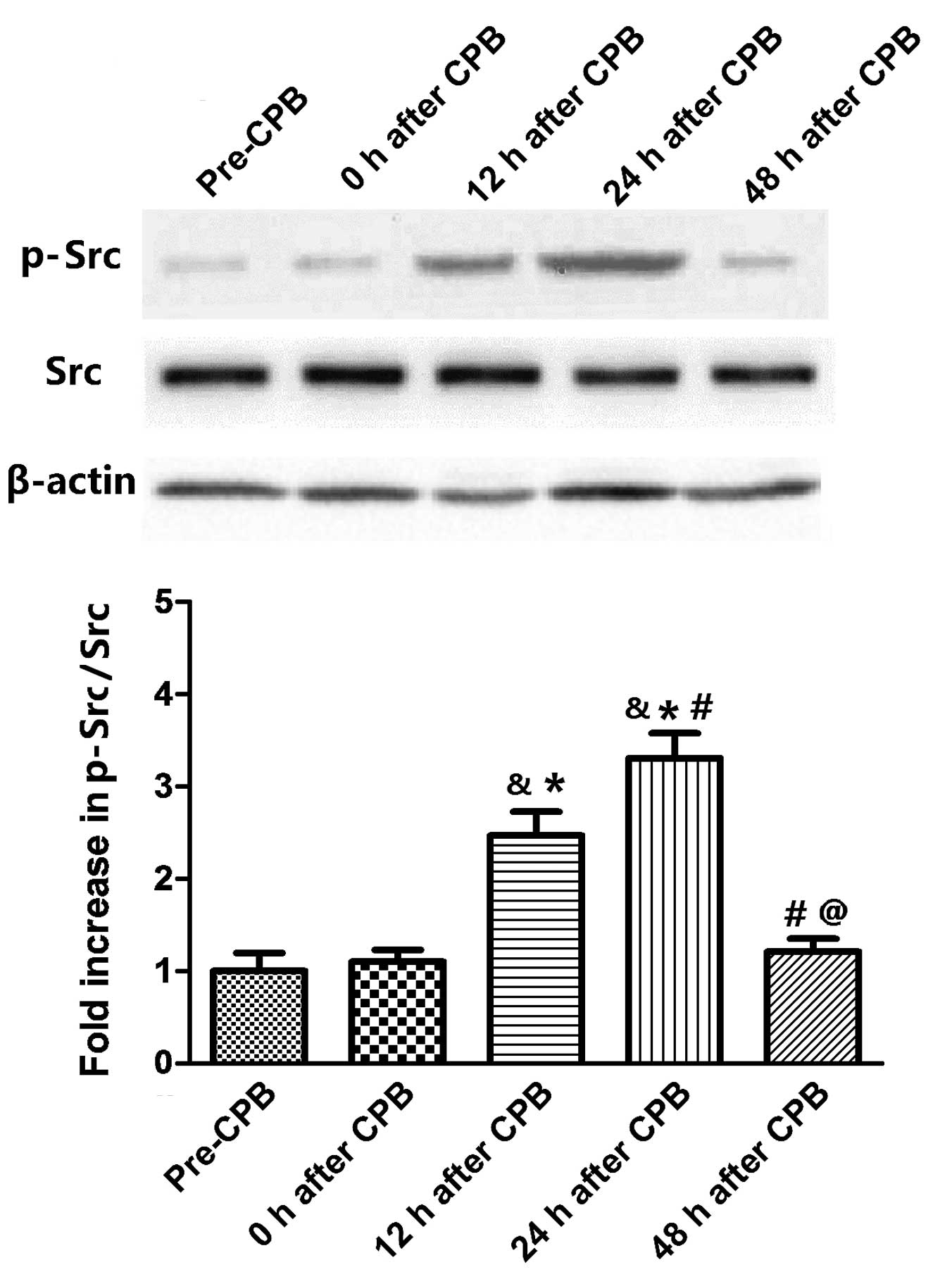
Src phosphorylation increases in the lung
tissues following CPB surgery
Fig. 3 reveals the
time course of Src phosphory-lation in lung tissues. Src
phosphorylation was not altered immediately following CPB surgery.
However, Src phos-phorylation increased at 12 h after CPB and
peaked at 24 h after CPB (P<0.05, compared with the 12 h after
CPB group). At 48 h after CPB, it regressed to the normal level
(P<0.05, compared with 12 and 24 h after CPB).
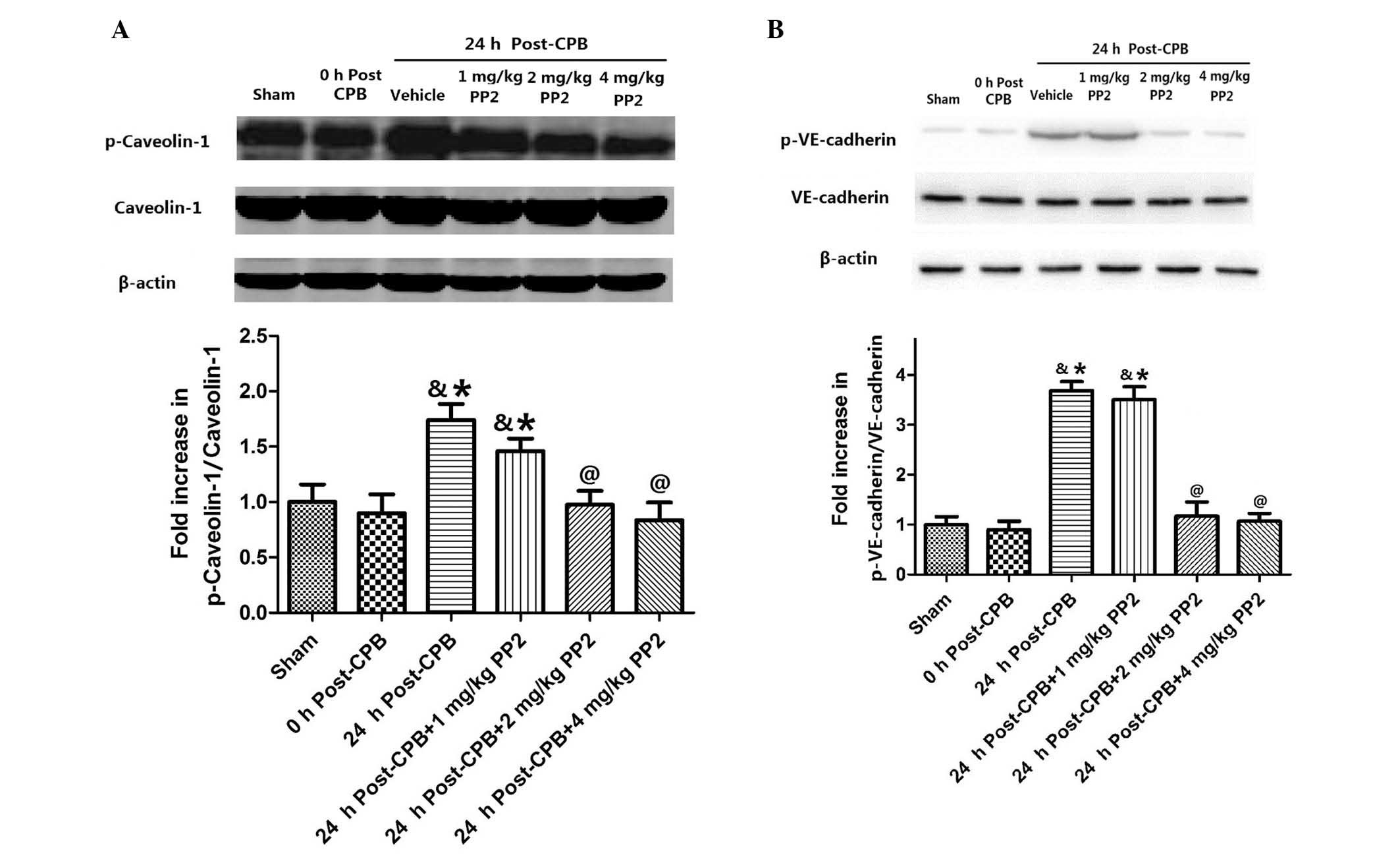
PP2 attenuates increases in caveolin-1
and VE-cadherin phosphorylation in the lung tissues following
CPB
In order to examine the effect of Src kinase
activation on caveolin-1 and VE-cadherin, caveolin-1 and
VE-cadherin phosphorylation in the lung tissues was measured
(Fig. 4). Caveolin-1
phosphorylation did not immediately change following CPB surgery,
but significantly increased (P<0.05, compared with the sham
group) at 24 h after CPB. Treatment with three doses of PP2 (1, 2
and 4 mg/kg) inhibited caveolin-1 phosphorylation in a
dose-dependent manner (Fig. 4A).
No significant difference was observed in VE-cadherin
phosphorylation between the sham group and at 0 h after CPB
surgery. However, at 24 h after CPB, VE-cadherin phosphorylation
significantly increased (P<0.05) compared with the sham group.
Treatment with high doses of PP2 (2 and 4 mg/kg) significantly
inhibited VE-cadherin phosphorylation (Fig. 4B).
Discussion
Acute lung injury is among the leading cause of
morbidity and mortality in patients who have undergone cardiac
surgery necessitating CPB (19,20).
Numerous factors may contribute to acute lung injury, including the
exposure of blood to the artificial surface of the CPB machine,
ischemia-reperfusion and lung ventilator-elicited inflammatory
reactions (21). Certain previous
studies have suggested that pulmonary microvascular permeability is
a major contributor to acute lung injury (22,23),
however, the mechanisms have not yet been investigated. In the
present study, the EBD results indicated impaired pulmonary
microvascular permeability following CPB surgery. The microvascular
permeability started to increase following CPB surgery and then
peaked 24 h later. CPB also induced significant increases in the
neutrophil count and TNF-α, IL-1β and IL-6 in BALF, indicating the
provoked inflammatory reaction and increased pulmonary
microvascular permeability. Neutrophils, TNF-α and IL-6 in BALF
reached a peak at 24 h post CPB, while IL-1β in BALF continued to
increase until 48 h post CPB. These data revealed the time course
of the inflammatory reaction, which emerged following CPB surgery
and peaked at 24–48 h after CPB surgery. The time course of these
changes correlates with the alterations in pulmonary microvascular
permeability.
Src kinases belong to the non-receptor tyrosine
kinase family, which contains c-Src, Fyn, Yes, Yrk, Blk, Fgr, Hck,
Lck and Lyn (24). In response to
stimulation of a variety of cell surface receptors, including
tyrosine kinase receptors, integrin receptors and G protein-coupled
receptors, the activity of Src can be upregulated by
phosphorylation at Tyr 416, located in the catalytic domain
(24). It has been demonstrated
that Src mediates vascular endothelial permeability responses to
TNF, reactive oxygen species, angiogenesis and vascular leakage
(25–27). Inhibition of the Src family reduces
cerebral edema and eradicates the increase in albumin permeability
caused by C5α-activated neutrophils in venules (28,29).
Neutrophil activation stimulates Src phosphorylation at Tyr 416 and
decreases phosphorylation at Tyr 527, which upregulates Src
activity (30). The results
demonstrated that Src phosphorylation (activation) accompanied the
increase in pulmonary microvascular leakage, while the
administration of PP2, an inhibitor of the Src kinase, attenuated
the alterations in pulmonary microvascular leakage, neutrophil
count and proinflammatory cytokines (with the exception of TNF-α)
in BALF caused by CPB, indicating that Src kinase has an important
role in the effects of CPB on pulmonary microvascular permeability.
For neutrophils and IL-6 in BALF, PP2 administration reduced their
values to a level equivalent to the sham group. However, the fact
that TNF-α was unaltered by PP2 suggests that the induction of
TNF-α may not be regulated by Src kinase.
Src kinase may regulate the microvascular
permeability and endothelial barrier structure through multiple
pathways, including mitogen-activated protein kinase, myosin light
chain kinase, β-catenin, or focal adhesion proteins (24,31,32).
The present study focused on caveolin-1 and VE-cadherin, two
important proteins in the regulation of pulmonary microvascular
permeability. Caveolae were originally identified as 50–100 nm
flask-shaped, non-clathrin-coated invaginations of the plasma
membrane, which are important in transendothelial vesicular
transport. Caveolin-1 is a critical protein for caveolae-mediated
endocytosis and transcytosis in endothelial cells (33). It contains a scaffolding domain and
acts as an inhibitory regulator of endothelial Rac1 signaling in
the regulation of endothelial permeability (34,35).
Tyrosine phosphorylation of caveolin-1 is important in the
pathogenesis of oxidant-induced pulmonary vascular
hyperpermeability (36). Previous
studies have demonstrated that an increase in transcellular
permeability was dependent on Src-mediated phosphorylation of
caveolin-1 (13,34,37,38).
The pulmonary vascular hyperpermeability induced by activation of
neutrophils adherent to the vessel wall is dependent on signaling
via caveolin-1 and increased caveolae-mediated transcytosis
(39). Sun et al
demonstrated that phosphorylation of caveolin-1 is an important
mechanism mediating oxidant-induced vascular hyperpermeability by
stimulating paracellular and caveolae-mediated transcellular
permeability (36). The results
from the present study demonstrated that caveolin-1 phosphorylation
was not altered immediately following CPB surgery, but was
significantly increased at 24 h after CPB. Treatment with 1 mg/kg
PP2 did not alter caveolin-1 phosphorylation, however, 2 and 4
mg/kg PP2 inhibited caveolin-1 phosphorylation, indicating that Src
kinase may function via the activation of caveolin-1.
VE-cadherin is a classical cadherin from the
cadherin family, which is critical in endothelial cell biology and
vascular permeability through homophilic binding to other
VE-cadherins expressed on adjacent endothelial cells (40). Numerous stimuli, including TNF and
vascular endothelial growth factor, may cause the phosphorylation
of VE-cadherin, in which Src kinase acts as a key pathway mediator
(14,41). It was also demonstrated that
proinflammatory cytokines could induce the phosphorylation of
VE-cadherin and the endocytosis of VE-cadherin in a
β-arrestin-dependent manner through the Src kinase pathway
(42). Src activation could also
cause the phosphorylation of VE-cadherin by stimulation of
H2O2 (43,44).
The present study demonstrated that VE-cadherin phosphorylation was
not altered immediately following CPB surgery, however, it
significantly increased at 24 h post CPB. VE-cadherin
phosphorylation was inhibited by treatment with PP2 (2 and 4
mg/kg). The results revealed that Src was involved in the
phosphorylation of VE-cadherin, however, whether caveolin-1
mediated this process or not remains to be elucidated. A previous
study demonstrated that the knockdown of caveolin-1 induced a
decrease in VE-cadherin localized at inter-endothelial junctions
(45). The interaction of
caveolin-1 and VE-cadherin activation in Src-mediated pulmonary
vascular hyperpermeability requires further investigation.
In conclusion, the present study demonstrated that
pulmonary microvascular permeability was increased following CPB
through the Src kinase pathway. The activation of caveolin-1 and
VE-cadherin appears to be the downstream effect of Src kinase
phosphorylation. Inhibition of this pathway may provide a potential
therapy for acute lung injury following cardiac surgery.
Acknowledgments
This study was funded by the Science and Technology
Commission of Shanghai Municipality (grant no. 11ZR1423700).
References
|
1
|
Gibbon JH Jr: Application of a mechanical
heart and lung apparatus to cardiac surgery. Minn Med. 37:171–185.
1954.PubMed/NCBI
|
|
2
|
Edmunds LH Jr: Advances in the heart-lung
machine after John and Mary Gibbon. Ann Thorac Surg.
76:S2220–S2223. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wahba A: Centrifugal blood pump use in
routine cardiac surgery. Interact Cardiovasc Thorac Surg.
5:299–300. 2006. View Article : Google Scholar
|
|
4
|
Cox CS Jr, Allen SJ and Brennan MS:
Analysis of intestinal microvascular permeability associated with
cardiopulmonary bypass. J Surg Res. 83:19–26. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ng CS, Wan S, Yim AP and Arifi AA:
Pulmonary dysfunction after cardiac surgery. Chest. 121:1269–1277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Serraf A, Aznag H, Baudet B, Détruit H,
Séccatore F, Mazmanian MG and Planché C: Pulmonary vascular
endothelial growth factor and nitric oxide interaction during total
cardiopulmonary bypass in neonatal pigs. J Thorac Cardiovasc Surg.
125:1050–1057. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Okutani D, Lodyga M, Han B and Liu M: Src
protein tyrosine kinase family and acute inflammatory responses. Am
J Physiol Lung Cell Mol Physiol. 291:L129–L141. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oyaizu T, Fung SY, Shiozaki A, Guan Z,
Zhang Q, dos Santos CC, Han B, Mura M, Keshavjee S and Liu M: Src
tyrosine kinase inhibition prevents pulmonary
ischemia-reperfusion-induced acute lung injury. Intensive Care Med.
38:894–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miyahara T, Hamanaka K, Weber DS, Drake
DA, Anghelescu M and Parker JC: Phosphoinositide 3-kinase, Src and
Akt modulate acute ventilation-induced vascular permeability
increases in mouse lungs. Am J Physiol Lung Cell Mol Physiol.
293:L11–L21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu YY, Li LF, Fu JY, Kao KC, Huang CC,
Chien Y, Liao YW, Chiou SH and Chang YL: Induced pluripotent stem
cell therapy ameliorates hyperoxia-augmented ventilator-induced
lung injury through suppressing the Src pathway. PLoS One.
9:e1099532014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anderson RG: The caveolae membrane system.
Annu Rev Biochem. 67:199–225. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cohen A, Hnasko R, Schubert W and Lisanti
MP: Role of caveolae and caveolins in health and disease. Physiol
Rev. 84:1341–1379. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Minshall RD, Tiruppathi C, Vogel SM and
Malik AB: Vesicle formation and trafficking in endothelial cells
and regulation of endothelial barrier function. Histochem Cell
Biol. 117:105–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dejana E, Orsenigo F and Lampugnani MG:
The role of adherens junctions and VE-cadherin in the control of
vascular permeability. J Cell Sci. 121:2115–2122. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Orsenigo F, Giampietro C, Ferrari A,
Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Koh GY,
Franco D, et al: Phosphorylation of VE-cadherin is modulated by
haemodynamic forces and contributes to the regulation of vascular
permeability in vivo. Nat Commun. 3:12082012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Institute of Laboratory Animal Resources
(US): Committee on Care, Use of Laboratory Animals, and National
Institutes of Health (US). Division of Research Resources: Guide
for the care and use of laboratory animals. 7th edition. National
Academies Press; Washington, DC: 1996
|
|
17
|
Zhu J, Yin R, Shao H, Dong G, Luo L and
Jing H: N-acetylcysteine to ameliorate acute renal injury in a rat
cardiopulmonary bypass model. J Thorac Cardiovasc Surg.
133:696–703. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cavriani G, Oliveira-Filho RM, Trezena AG,
da Silva ZL, Domingos HV, de Arruda MJ, Jancar S and Tavares de
Lima W: Lung microvascular permeability and neutrophil recruitment
are differently regulated by nitric oxide in a rat model of
intestinal ischemia-reperfusion. Eur J Pharmacol. 494:241–249.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Altmay E, Karaca P, Yurtseven N, Ozkul V,
Aksoy T, Ozler A and Canik S: Continuous positive airway pressure
does not improve lung function after cardiac surgery. Can J
Anaesth. 53:919–925. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Apostolakis E, Filos KS, Koletsis E and
Dougenis D: Lung dysfunction following cardiopulmonary bypass. J
Card Surg. 25:47–55. 2010. View Article : Google Scholar
|
|
21
|
Engels M, Bilgic E, Pinto A, Vasquez E,
Wollschläger L, Steinbrenner H, Kellermann K, Akhyari P,
Lichtenberg A and Boeken U: A cardiopulmonary bypass with deep
hypothermic circulatory arrest rat model for the investigation of
the systemic inflammation response and induced organ damage. J
Inflamm (Lond). 11:262014. View Article : Google Scholar
|
|
22
|
Macnaughton PD, Braude S, Hunter DN,
Denison DM and Evans TW: Changes in lung function and pulmonary
microvascular permeability after cardiopulmonary bypass. Crit Care
Med. 20:1289–1294. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Messent M, Sinclair DG, Quinlan GJ, Mumby
SE, Gutteridge JM and Evans TW: Pulmonary vascular permeability
after cardiopulmonary bypass and its relationship to oxidative
stress. Crit Care Med. 25:425–429. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thomas SM and Brugge JS: Cellular
functions regulated by Src family kinases. Annu Rev Cell Dev Biol.
13:513–609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kevil CG, Okayama N and Alexander JS:
H(2)O(2)-mediated permeability II: Importance of tyrosine
phosphatase and kinase activity. Am J Physiol Cell Physiol.
281:C1940–C1947. 2001.PubMed/NCBI
|
|
26
|
Nwariaku FE, Liu Z, Zhu X, Turnage RH,
Sarosi GA and Terada LS: Tyrosine phosphorylation of vascular
endothelial cadherin and the regulation of microvascular
permeability. Surgery. 132:180–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eliceiri BP, Paul R, Schwartzberg PL, Hood
JD, Leng J and Cheresh DA: Selective requirement for Src kinases
during VEGF-induced angiogenesis and vascular permeability. Mol
Cell. 4:915–924. 1999. View Article : Google Scholar
|
|
28
|
Paul R, Zhang ZG, Eliceiri BP, Jiang Q,
Boccia AD, Zhang RL, Chopp M and Cheresh DA: Src deficiency or
blockade of Src activity in mice provides cerebral protection
following stroke. Nat Med. 7:222–227. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tinsley JH, Ustinova EE, Xu W and Yuan SY:
Src-dependent, neutrophil-mediated vascular hyperpermeability and
beta-catenin modification. Am J Physiol Cell Physiol.
283:1745–1751. 2002. View Article : Google Scholar
|
|
30
|
Yuan SY: Protein kinase signaling in the
modulation of microvascular permeability. Vascul Pharmacol.
39:213–223. 2002. View Article : Google Scholar
|
|
31
|
Eliceiri BP, Puente XS, Hood JD, Stupack
DG, Schlaepfer DD, Huang XZ, Sheppard D and Cheresh DA:
Src-mediated coupling of focal adhesion kinase to integrin
alpha(v)beta5 in vascular endothelial growth factor signaling. J
Cell Biol. 157:149–160. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shi S, Garcia JG, Roy S, Parinandi NL and
Natarajan V: Involvement of c-Src in diperoxovanadate-induced
endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol
Physiol. 279:L441–L451. 2000.PubMed/NCBI
|
|
33
|
Drab M, Verkade P, Elger M, Kasper M, Lohn
M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al:
Loss of caveolae, vascular dysfunction and pulmonary defects in
caveolin-1 gene-disrupted mice. Science. 293:2449–2452. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Minshall RD, Sessa WC, Stan RV, Anderson
RG and Malik AB: Caveolin regulation of endothelial function. Am J
Physiol Lung Cell Mol Physiol. 285:L1179–L1183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gonzalez E, Nagiel A, Lin AJ, Golan DE and
Michel T: Small interfering RNA-mediated down-regulation of
caveolin-1 differentially modulates signaling pathways in
endothelial cells. J Biol Chem. 279:40659–40669. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun Y, Hu G, Zhang X and Minshall RD:
Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary
vascular permeability via paracellular and transcellular pathways.
Circ Res. 105:676–685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shajahan AN, Timblin BK, Sandoval R,
Tiruppathi C, Malik AB and Minshall RD: Role of Src-induced
dynamin-2 phosphorylation in caveolae-mediated endocytosis in
endothelial cells. J Biol Chem. 279:20392–20400. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Minshall RD, Tiruppathi C, Vogel SM, Niles
WD, Gilchrist A, Hamm HE and Malik AB: Endothelial cell-surface
gp60 activates vesicle formation and trafficking via Gi-coupled Src
kinase signaling pathway. J Cell Biol. 150:1057–1070. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu G, Vogel SM, Schwartz DE, Malik AB and
Minshall RD: Intercellular adhesion molecule-1-dependent neutrophil
adhesion to endothelial cells induces caveolae-mediated pulmonary
vascular hyperpermeability. Circ Res. 102:e120–e131. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lim MJ, Chiang ET, Hechtman HB and Shepro
D: Inflammation-induced subcellular redistribution of VE-cadherin,
actin and gamma-catenin in cultured human lung microvessel
endothelial cells. Microvasc Res. 62:366–382. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gavard J, Hou X, Qu Y, Masedunskas A,
Martin D, Weigert R, Li X and Gutkind JS: A role for a
CXCR2/phosphatidylinositol 3-kinase gamma signaling axis in acute
and chronic vascular permeability. Mol Cell Biol. 29:2469–2480.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gavard J and Gutkind JS: VEGF controls
endothelial-cell permeability by promoting the
beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol.
8:1223–1234. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mehta D and Malik AB: Signaling mechanisms
regulating endothelial permeability. Physiol Rev. 86:279–367. 2006.
View Article : Google Scholar
|
|
44
|
Aberle H, Schwartz HR and Kemler R:
Cadherin-catenin complex: Protein interactions and their
implications for cadherin function. J Cell Biochem. 61:514–523.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Song L, Ge S and Pachter JS: Caveolin-1
regulates expression of junction-associated proteins in brain
microvascular endothelial cells. Blood. 109:1515–1523. 2007.
View Article : Google Scholar
|