Introduction
Melanoma is a lethal skin cancer with poor prognosis
due to its capacity for early stage metastasis and resistance to
chemotherapy, with only 14% of patients with metastatic melanoma
surviving for 5 years (1). There
is an urgent demand for the development of higher efficacy
treatment strategies, and the elucidation of the associated
molecular mechanisms.
Nuclear factor-κB (NF-κB) is an essential regulator
of gene transcription for numerous genes, including those that are
critically involved in apoptosis; and the aberrant regulation of
NF-κB an important mechanism involved in the development of
melanoma. Currently, the anti-apoptotic functions of NF-κB are
suggested to be predominantly mediated by the upregulation of
anti-apoptotic genes including cellular Fas-associated death
domain-like interleukin-1β-converting enzyme (FLICE)-inhibitory
protein (c-FLIP), members of the Bcl-2 family and
X-chromosome-linked inhibitor of apoptosis (2). In addition to these, recent studies
have shown that NF-κB suppresses tumor necrosis factor
(TNF)-α-induced cell death by inhibiting prolonged c-Jun N-terminal
kinase (JNK) activation (3).
However, the detailed molecular mechanisms remain to be fully
elucidated.
c-FLIP regulates death receptor-mediated apoptosis
(4) and inhibits cluster of
differentiation (CD)95- and TNF-related apoptosis-inducing ligand
receptor (TRAIL-R)-mediated apoptosis by interfering with the
activation of caspase-8 (5). It is
expressed as long (c-FLIPL), short (c-FLIPS)
and Raji (c-FLIPR) splice variants in human cells
(6). Particularly,
c-FLIPL and c-FLIPS are known to have
multifunctional roles in various signaling pathways, including
activating and/or upregulating several molecules involved in
cytoprotective signaling (7).
In the present study, eukaryotic short hairpin
(sh)RNA expression vectors for c-FLIP isoforms were successfully
cloned, and their role in cellular proliferation was investigated.
In addition, the effect of c-FLIPL on JNK signaling was
investigated.
Materials and methods
Reagents and cell culture
The following antibodies were used and purchased as
indicated: Polyclonal rabbit anti-c-FLIP (1:500; NF6; Enzo Life
Sciences, Inc., Farmingdale, NY, USA); polyclonal rabbit
anti-phosphorylated (p)-stress activated protein kinase (SAPK)/JNK
(Thr183/Tyr185) (1:1,000; 9251; Cell Signaling Technology, Inc.,
Danvers, MA, USA); polyclonal rabbit anti-SAPK/JNK (1:1,000; 9252;
Cell Signaling Technology, Inc.); monoclonal mouse anti-β-actin
(1:2,000; sc-47778; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA). TNF-α was purchased from Sigma-Aldrich (St. Louis, MO,
USA).
The A375, A875 and SK-Mel-1 cell lines were
purchased from Shanghai Maisha Biotechnology Co., Ltd. (Shanghai,
China). The SK-Mel-28 cell line was kindly provided by Dr Peter M.
Blumberg (National Cancer Institute, Bethesda, MD, USA). Cell lines
were cultured with Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a
humid environment with 5% CO2.
Plasmid constructs for human c-FLIP
shRNAs
To construct shRNAs targeting human c-FLIP (long
form accession no. U97074; short form accession no. U97075), the
following oligonucleotides were designed according to Tushul's
principle (8,9). Pgenesil-1 (10) (Wuhan Genesil Biotechnology Co.,
Ltd., Wuhan, China) containing complementary DNA of green
fluorescent protein and the kanamycin-resistance gene was used for
the vector backbone. The control-scrambled shRNA was constructed by
the insertion of a similar structure but encoding a nonsense
minigene with no homology to any known sequences in human and mouse
genomes. Recombinant chimeric plasmids were verified by restriction
enzyme analysis and sequencing, and constructs that targets the
both isoforms, the long isoform and the short isoform were named as
c-FLIP shRNA (target sequence 5′-AACTGCTCTACAGAGTGAGGC-3′),
c-FLIPL shRNA (target sequence
5′-AAGATGAAGAGCAAGCCCCTA-3′) and c-FLIPS shRNA (target
sequence 5′-ATGCCCATTGTCCTGATCTGA-3′), respectively. A control
scrambled shRNA (target sequence 5′-AATTCTCCGAACGTGTCACGT-3′) was
also designed.
Transient transfection of c-FLIP
shRNAs
c-FLIP, c-FLIPL and c-FLIPS
shRNAs were transfected into A875 cells using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Cells were used for subsequent
experiments 48 h following transfection. TNF-α (10 ng/ml) was added
to cells (5×104 cells/ml) for 4 or 8 h.
Western blotting
Cells (5×105) were lysed in
radioimmu-noprecipitation buffer (50 mM Tris-HCl, pH 8.0), 150 mM
NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% sodium dodecyl
sulfate, 25 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM
sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 1 mg/ml
aprotinin and 1 mg/ml leupeptin), the lysate was then centrifuged
at 12,000 × g for 15 min at 4°C, the supernatant was collected for
western blot analysis as described previously (11). Briefly, protein was separated on
10% SDS-Page gel (Wuhan Boster Biological Technology, Ltd., Wuhan,
China) and transferred onto a polyvinylidene difluoride membrane
(Pall Corporation, Port Washington NY, USA). Then, 5% skimmed milk
was used to block the membrane for 1 h at room temperature. The
membrane was then incubated with primary antibodies (listed under
reagents) at 4°C overnight, and washed with phosphate buffered
saline with Tween-20 (PBST; Beijing Dingguo Biotechnology Co.,
Ltd., Beijing, China) 5 times for 10 min. Then, the membrane was
incubated with the following secondary antibodies at room
temperature for 1 h: Horseradish peroxidase (HRP)-conjugated donkey
anti-rabbit (1:5,000; sc-2313), or donkey anti-mouse HRP-conjugated
mouse antibodies (1:5,000; sc-2314) (Santa Cruz Biotechnology,
Inc.). Finally, the membranes were washed with PBST 5 times for 10
min. Immunoreactivity was measured using enhanced chemiluminescence
(BeyoECL Plus kit; Beyotime Institute of Biotechnology, Shanghai,
China), and the band density was analyzed using ImagePro Plus
software (version 6.0; Media Cybernetics, Inc., Rockville, MD,
USA).
Determination of cell viability
Cell viability was determined by 3-(4,5)-dimethylthiahiazol-2-y1]-2,5-diphenyltetrazolium
bromide (MTT) assay, using a kit purchased from Roche Diagnostics
GmbH (Mannheim, Germany). Cells (5×103) were plated in
96-well plates and incubated for 48 h and assayed for the number of
viable cells according to the manufacturer's instructions. The
substrate color development was monitored at 570 nm using a
microplate spectrometer (BioTek Synergy HT Multi-Mode Microplate
Reader; Bio-Tek Instruments, Inc., Winooski, VT, USA). The assay
was repeated three times.
Statistical analysis
Data analysis was performed using SPSS software,
version 17.0 (SPSS, Inc., Chicago, IL, USA). Differences between
groups were analyzed using one-way analysis of variance, followed
by Tukey's multiple comparisons test to identify differences
between specific groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
c-FLIP expression in melanoma cell
lines
Abundant expression of c-FLIP protein was observed
in the A375, A875, SK-Mel-1 and SK-Mel-28 melanoma cell lines, as
determined by western blotting. The A875 cell line, which had the
highest expression of c-FLIP among the melanoma cell lines tested,
was selected for further experiments (Fig. 1).
Silencing c-FLIP in A875 cells by c-FLIP
shRNAs
The knockdown efficiency of c-FLIP shRNAs was
subsequently investigated. The shRNA constructs were transfected
individually into A875 cells, and c-FLIP, c-FLIPL and
c-FLIPS shRNAs all efficiently suppressed the expression
of c-FLIP, as shown in Fig. 2.
c-FLIPL and c-FLIPS
shRNA inhibit cell proliferation
A previous study observed a higher rate of apoptosis
in A875 cells transfected with the long or short form of c-FLIP
siRNA compared with non-transfected cells or cells transfected with
control siRNA. However, the difference between the A875 cells
transfected with c-FLIPL and c-FLIPS siRNA
was not significant (12). In the
present study, alterations in cellular proliferation in A875 cells
transfected with c-FLIPL shRNA, c-FLIPS
shRNA, scrambled shRNA and untransfected A875 cells were detected
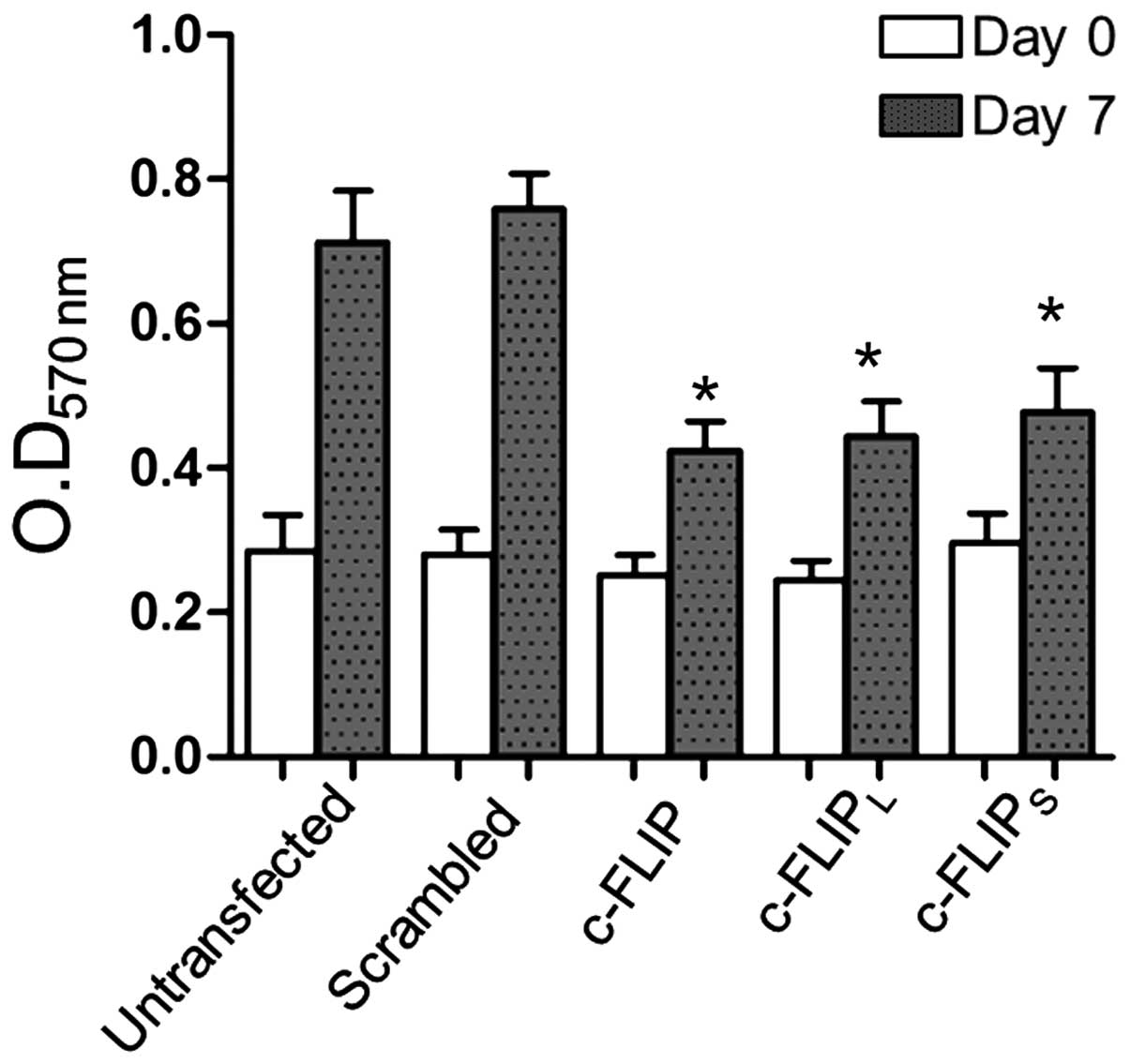
by MTT assay. Cells were cultured for 7 days following
transfection, the optical density values were compared. The optical
density of A875 cells transfected with the c-FLIP,
c-FLIPL and c-FLIPS shRNA eukaryotic
expression vectors were observed to be significantly reduced
compared with the untransfected A875 cells and scrambled
shRNA-transfected cells (Fig. 3),
suggesting a diminished capacity for cell proliferation. In
addition, no significant differences were observed between the
c-FLIP, c-FLIPL and c-FLIPS shRNA transfected
groups.
TNF-α induces prolonged JNK activation in
A875 cells transfected with c-FLIP shRNA
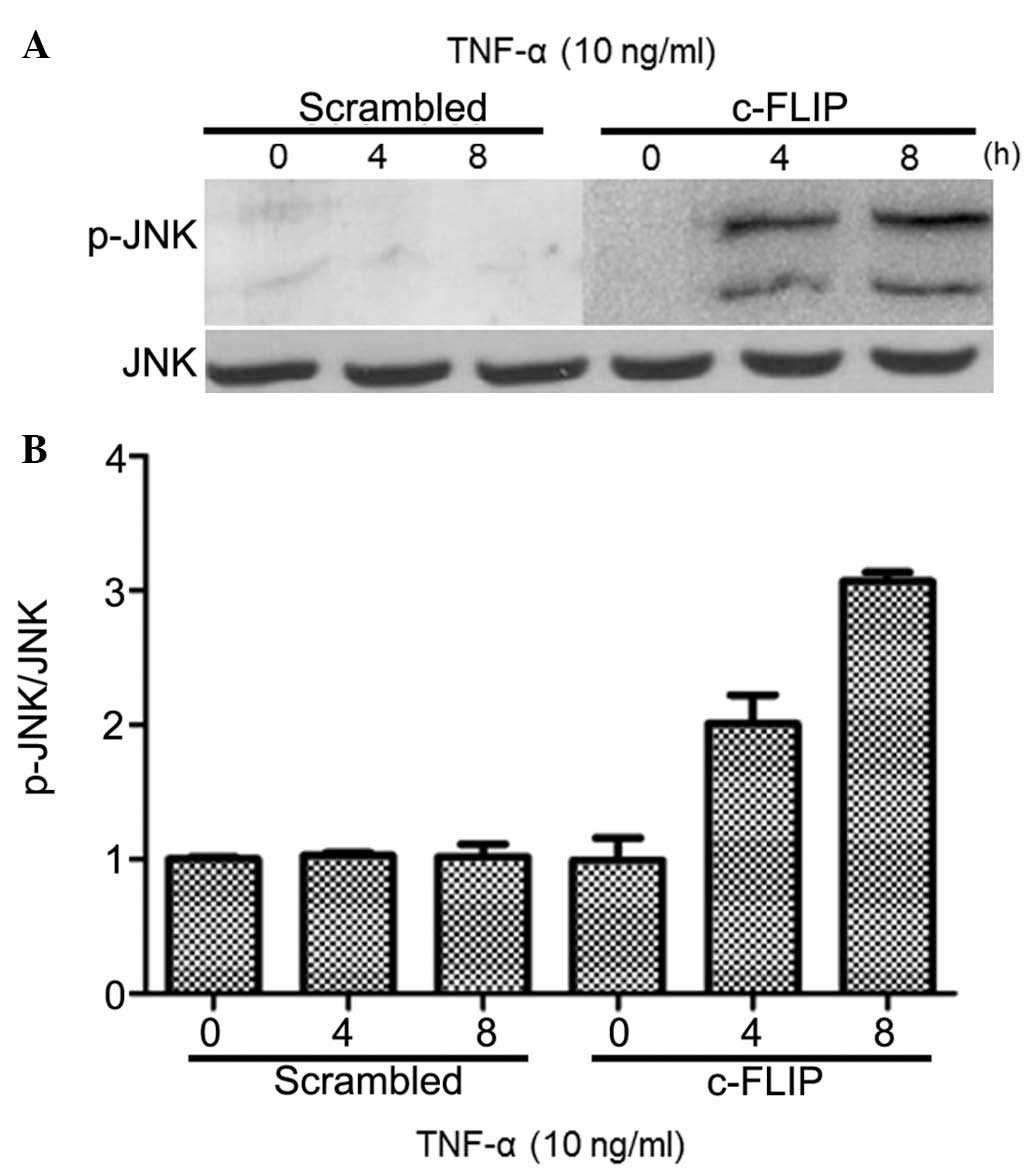
c-FLIP is a downstream molecule of TNF-α. and the
effect of c-FLIP inhibition on apoptosis, induced by TNF-α, was
analyzed. A875 cells growing in the logarithmic phase were
transfected with c-FLIP shRNA to target both isoforms, and treated
with TNF-α (10 ng/ml) for 0, 4 and 8 h. Transfection with c-FLIP
shRNA induced JNK activation upon treatment with TNF-α, and this
activation that was markedly prolonged up to 8 h following
treatment (Fig. 4). JNK activation
was not observed in cells transfected with scrambled shRNA. These
results suggested that increased c-FLIP expression may inhibit JNK
activation in A875 cell line.
TNF-α induces prolonged JNK activation in
A875 cells transfected with c-FLIPL shRNA but not c-FLIPS
shRNA
Next, cells were transfected with c-FLIPL
and c-FLIPS shRNAs, and subsequently treated the cells
with TNF-α (10 ng/ml). As shown in Fig. 5, TNF-α induced JNK activation in
A875 cells transfected with c-FLIPL shRNA, however, not
in those transfected with c-FLIPS shRNA. This suggests
that only c-FLIPL shRNA is able to induce JNK activation
in A875 cell lines, although both c-FLIPL and
c-FLIPS shRNA are able to suppress c-FLIP
expression.
Discussion
The expression of c-FLIP has been demonstrated to be
a factor involved in the resistance of tumor cells to death ligands
such as FasL and TRAIL, and certain reports have demonstrated that
downregulation of c-FLIP results in the resensitizing of various
types of resistant tumor cells (13–16).
These previous studies have delineated the multifunctional role of
c-FLIP in diverse signaling pathways that regulate apoptosis,
proliferation, carcinogenesis, and the survival of cancer cells
(17,18).
The JNK pathway represents a subgroup of mitogen
activated protein kinases (MAPK) that are primarily activated by
cytokines and environmental stresses (19). Specific stimuli trigger the
activation of MAP3Ks, which then phosphorylate and activate the
MAP2K isoforms, MKK4 and MKK7, and subsequently phosphorylate and
activate JNK (20). JNKs belong to
the superfamily of MAPKs that are involved in the regulation of
cell proliferation, differentiation and apoptosis (21). Analysis of the pathways regulated
by JNKs have indicated that JNKs are indispensable for both cell
proliferation and apoptosis (22).
Whether the activation of JNKs leads to cell proliferation or
apoptosis is dependent on the stimuli and the cell type involved in
such activations (23). There is
currently few studies reporting on the regulatory mechanisms of JNK
in melanoma. Jørgensen et al (24) analyzed sections from 154 primary
(93 superficial and 61 nodular melanomas) and 73 metastatic
melanomas, together with 34 benign samples using
immunohistochemistry. They observed that 35% of the primary and 25%
of the metastatic melanoma samples expressed variable levels of
p-JNK. By contrast, 73.5% of the benign samples expressed p-JNK
(24). Numerous previous studies
have indicated that c-FLIP-specific siRNA can induce apoptosis in
breast, colorectal, lung and prostate cancer cell lines (25–28).
A previous study indicated that c-FLIPL enhances tumor
cell viability via the activation of extracellular signal-regulated
kinase and focal adhesion kinase signaling (29). In addition, previous studies have
indicated that overexpression of c-FLIPL prevented the
proteasomal degradation of β-catenin, thus increasing the β-catenin
levels induced by Wnt signaling or cyclin D, thereby promoting
tumor cell proliferation and cell cycle progression (30,31).
Numerous reports have suggested that the overexpression of
c-FLIPL promotes tumorigenesis and the invasion of
endometrial cancer and cervical cancer (32,33).
These studies indicated that c-FLIP has multiple functions in
signaling pathways regulating apoptosis, proliferation,
tumorigenesis and tumor cell survival. In order to verify whether
c-FLIP expression has a regulatory role in the JNK signaling
pathway, the expression of c-FLIP was measured in several malignant
melanoma cell lines, with A875 cells observed to have the greatest
expression and therefore was selected for further experiments. The
present study demonstrated that knockdown of c-FLIP or
c-FLIPL however, not c-FLIPS induced
prolonged JNK activation, which was consistent with previous
studies (34,35). A previous study reported that
down-regulation of c-FLIP expression was able to induce apoptosis
in A875 cells (12). In the
present study, using A875 melanoma cells, the effects of c-FLIP,
c-FLIPL and c-FLIPS shRNA on the biological
characteristics of cells were observed, indicating that different
subtypes of c-FLIP shRNA were able to inhibit tumor cell
proliferation. These results suggest that high expression of c-FLIP
in melanoma cells may not only provide cells with resistance to
apoptosis, but also promote cancer cell growth and proliferation.
In the current study, both c-FLIPL and
c-FLIPS shRNAs demonstrated a marked effect on cell
proliferation, however, c-FLIPS shRNA did not show any
effect on JNK activation. This suggests that c-FLIPS may
inhibit cellular proliferation though mechanisms other than JNK
activation, highlighting some unsolved mechanistic questions for
further research.
To date, several small molecules have been found to
lower c-FLIP expression and to sensitize tumor cells to
death-receptor mediated apoptosis (4). In addition, the inhibitors of several
kinases (MAP/ERK kinase1/2, protein kinase C and phosphoinositide
3-kinase) are reported to lower FLIP expression through blocking
the signaling pathways for FLIP transcription. We propose that
RNAi-targeted therapies towards key regulatory genes such as c-FLIP
will prove useful as a mono-therapy to treat certain types of
cancer, in addition to being used in combination with chemotherapy,
cytokine therapy or radiotherapy to sensitize drug resistant types
of cancer, such as melanoma.
In summary, the present study indicated that
c-FLIPL serves a role in melanoma proliferation via the
JNK pathway. The concept of transfecting a c-FLIP shRNA expression
vector to block c-FLIP holds promise as a clinical gene therapy
approach for melanoma.
Acknowledgments
This present study was supported by the Fund for
Technology Research and Development of Zhengzhou City (grant no.
141PPTGG319).
References
|
1
|
Miller AJ and Mihm MC Jr: Melanoma. N Engl
J Med. 355:51–65. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Karin M and Lin A: NF-kappaB at the
crossroads of life and death. Nat Immunol. 3:221–227. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kamata H, Honda S, Maeda S, Chang L,
Hirata H and Karin M: Reactive oxygen species promote
TNFalpha-induced death and sustained JNK activation by inhibiting
MAP kinase phosphatases. Cell. 120:649–661. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kataoka T: The caspase-8 modulator c-FLIP.
Crit Rev Immunol. 25:31–58. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang DW, Xing Z, Pan Y,
Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME and Yang
X: c-FLIP(L) is a dual function regulator for caspase-8 activation
and CD95-mediated apoptosis. EMBO J. 21:3704–3714. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Safa AR: c-FLIP, a master anti-apoptotic
regulator. Exp Oncol. 34:176–184. 2012.PubMed/NCBI
|
|
7
|
Safa AR and Pollok KE: Targeting the
anti-apoptotic protein c-FLIP for cancer therapy. Cancers (Basel).
3:1639–1671. 2011. View Article : Google Scholar
|
|
8
|
Wall NR and Shi Y: Small RNA: Can RNA
interference be exploited for therapy? Lancet. 362:1401–1403. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Medarova Z, Pham W, Farrar C, Petkova V
and Moore A: In vivo imaging of siRNA delivery and silencing in
tumors. Nat Med. 13:372–377. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen HL, Xu W, Wu ZY, Zhou LL, Qin RJ and
Tang HR: Vector-based RNAi approach to isoform-specific
down-regulation of vascular endothelial growth factor (VEGF) 165
expression in human leukemia cells. Leuk Res. 31:515–521. 2007.
View Article : Google Scholar
|
|
11
|
Sakon S, Xue X, Takekawa M, Sasazuki T,
Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T and Nakano
H: NF-kappaB inhibits TNF-induced accumulation of ROS that mediate
prolonged MAPK activation and necrotic cell death. EMBO J.
22:3898–3909. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian F, Lu JJ, Wang L, Li L, Yang J, Li Y,
Liu YQ, Shen GX, Tu YT and Tao J: Expression of c-FLIP in malignant
melanoma and its relationship with the clinicopathological features
of the disease. Clin Exp Dermatol. 37:259–265. 2012. View Article : Google Scholar
|
|
13
|
Mori T, Doi R, Toyoda E, Koizumi M, Ito D,
Kami K, Kida A, Masui T, Kawaguchi Y and Fujimoto K: Regulation of
the resistance to TRAIL-induced apoptosis as a new strategy for
pancreatic cancer. Surgery. 138:71–77. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dutton A, O'Neil JD, Milner AE, Reynolds
GM, Starczynski J, Crocker J, Young LS and Murray PG: Expression of
the cellular FLICE-inhibitory protein (c-FLIP) protects Hodgkin's
lymphoma cells from autonomous Fas-mediated death. Proc Natl Acad
Sci USA. 101:6611–6616. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abedini MR, Qiu Q, Yan X and Tsang BK:
Possible role of FLICE-like inhibitory protein (FLIP) in
chemoresistant ovarian cancer cells in vitro. Oncogene.
23:6997–7004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mezzanzanica D, Balladore E, Turatti F,
Luison E, Alberti P, Bagnoli M, Figini M, Mazzoni A, Raspagliesi F,
Oggionni M, et al: CD95-mediated apoptosis is impaired at receptor
level by cellular FLICE-inhibitory protein (long form) in wild-type
p53 human ovarian carcinoma. Clin Cancer Res. 10:5202–5214. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Safa AR: Roles of c-FLIP in apoptosis,
necroptosis, and autophagy. J Carcinog Mutagen. (Suppl 6):
0032013.PubMed/NCBI
|
|
18
|
Goldar S, Khaniani MS, Derakhshan SM and
Baradaran B: Molecular mechanisms of apoptosis and roles in cancer
development and treatment. Asian Pac J Cancer Prev. 16:2129–2144.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weston CR and Davis RJ: The JNK signal
transduction pathway. Curr Opin Cell Biol. 19:142–149. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar :
|
|
22
|
Uzdensky AB, Demyanenko SV and Bibov MY:
Signal transduction in human cutaneous melanoma and target drugs.
Curr Cancer Drug Targets. 13:843–866. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dhanasekaran DN and Reddy EP: JNK
signaling in apoptosis. Oncogene. 27:6245–6251. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jørgensen K, Davidson B and Flørenes VA:
Activation of c-jun N-terminal kinase is associated with cell
proliferation and shorter relapse-free period in superficial
spreading malignant melanoma. Mod Pathol. 19:1446–1455.
2006.PubMed/NCBI
|
|
25
|
Day TW, Huang S and Safa AR: C-FLIP
knockdown induces ligand-independent DR5-, FADD-, caspase-8- and
caspase-9-dependent apoptosis in breast cancer cells. Biochem
Pharmacol. 76:1694–1704. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sharp DA, Lawrence DA and Ashkenazi A:
Selective knockdown of the long variant of cellular FLICE
inhibitory protein augments death receptor-mediated caspase-8
activation and apoptosis. J Biol Chem. 280:19401–19409. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wilson TR, McLaughlin KM, McEwan M, Sakai
H, Rogers KM, Redmond KM, Johnston PG and Longley DB: C-FLIP: A key
regulator of colorectal cancer cell death. Cancer Res.
67:5754–5762. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wilson C, Wilson T, Johnston PG, Longley
DB and Waugh DJ: Interleukin-8 signaling attenuates TRAIL- and
chemotherapy-induced apoptosis through transcriptional regulation
of c-FLIP in prostate cancer cells. Mol Cancer Ther. 7:2649–2661.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park D, Shim E, Kim Y, Kim YM, Lee H, Choe
J, Kang D, Lee YS and Jeoung D: C-FLIP promotes the motility of
cancer cells by activating FAK and ERK and increasing MMP-9
expression. Mol Cells. 25:184–195. 2008.PubMed/NCBI
|
|
30
|
Naito M, Katayama R, Ishioka T, Suga A,
Takubo K, Nanjo M, Hashimoto C, Taira M, Takada S, Takada R, et al:
Cellular FLIP inhibits beta-catenin ubiquitylation and enhances Wnt
signaling. Mol Cell Biol. 24:8418–8427. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimada K, Nakamura M, Ishida E, Kishi M,
Matsuyoshi S and Konishi N: The molecular mechanism of
sensitization to Fas-mediated apoptosis by 2-methoxyestradiol in
PC3 prostate cancer cells. Mol Carcinog. 39:1–9. 2004. View Article : Google Scholar
|
|
32
|
Wang W, Wang S, Song X, Sima N, Xu X, Luo
A, Chen G, Deng D, Xu Q, Meng L, et al: The relationship between
c-FLIP expression and human papillomavirus E2 gene disruption in
cervical carcinogenesis. Gynecol Oncol. 105:571–577. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen HX, Liu YJ, Zhou XD and Luo RY:
Expression of cellular FLICE/caspase-8 inhibitory protein is
associated with malignant potential in endometrial carcinoma. Int J
Gynecol Cancer. 15:663–670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakajima A, Komazawa-Sakon S, Takekawa M,
Sasazuki T, Yeh WC, Yagita H, Okumura K and Nakano H: An
antiapoptotic protein, c-FLIPL, directly binds to MKK7 and inhibits
the JNK pathway. EMBO J. 25:5549–5559. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nakajima A, Kojima Y, Nakayama M, Yagita
H, Okumura K and Nakano H: Downregulation of c-FLIP promotes
caspase-dependent JNK activation and reactive oxygen species
accumulation in tumor cells. Oncogene. 27:76–84. 2008. View Article : Google Scholar
|