Introduction
Leukemia has the highest incidence rate
(3–4/100,000) among all types of pediatric cancer (patient age,
<18 years), and its incidence is increasing (1). In China, ~15,000 patients are newly
diagnosed with pediatric leukemia each year, of which >90%
classify as acute leukemia (AL). The development of AL is a
complex, multi-step process. Although combined chemotherapy and
hematopoietic stem cell transplantation technology have
considerably improved the survival rate of patients with pediatric
leukemia, the rate of recurrences in locations including the bone
marrow, testicles and central nervous system, is 25–30% (2). Further study of the pathogenesis of
leukemia will aid in the discovery of novel treatments and
prognostic markers.
For the past two decades, studies on the molecular
mechanisms of leukemia have mainly focused on chromosomal
abnormalities and protein-coding genes (3). Recently, non-coding microRNAs (miRNA)
were found to have promoting or suppressive effects on factors
associated with the occurrence, development, clinical manifestation
and prognosis of leukemia (4).
miRNAs are a class of endogenous, single-stranded, small,
non-coding RNA molecules containing 21–25 nucleotides. miRNAs are
thought to be generated through a selective amplification mechanism
and participate in a broad range of biological processes, including
ontogeny, cell differentiation, proliferation, apoptosis, aging and
stress (5–7). miRNA-expressing genes are often
clustered in fragile chromosome sites or cancer-associated genomic
regions (8). Abnormal expression
levels of miRNA in tumor cells are associated with tumor
occurrence, development and prognosis (9). It is known that miRNA expression is
regulated by DNA methylation and other epigenetic factors, which
may have a feedback interaction (10). As an important epigenetic
phenomenon, DNA methylation activity is frequently deregulated in
tumor cells and CpG island hypermethylation in tumor suppressor
genes may silence gene expression (11). Expression of miRNA is also
regulated by DNA methylation and other epigenetic factors, among
which miRNA itself may also affect DNA methylation. DNA
hypermethylation decreases the expression of tumor suppressor
miRNAs and increases the expression of oncogenic miRNAs. In
addition, histone modifications may also affect the expression of
miRNAs and cause tumor formation (12–15).
However, the epigenetic regulation of miRNAs in cancer has largely
remained elusive.
miR-34b belongs to the miR-34 family, which
comprises miR-34a, -b and -c. In humans, two gene clusters encode
miR-34, including the miR-34a gene located on chromosome 1p36 and
the miR-34b/c gene located on chromosome 11q23 (16). It has been found that miR-34b is
abnormally expressed in a variety of malignant cancers. In
colorectal cancer (5,17,18)
and gastric cancer (19,20), the CpG island in the miR-34b
promoter region is hypermethylated and the expression of miR-34b is
downregulated, which reduces its availability to exert its
tumor-suppressive function. In pancreatic cancer, miR-34b functions
as a tumor suppressor by targeting oncogene Smad3, and its low
expression is positively correlated with the tumor-nodes-metastasis
stage, lymph node metastasis and overall survival (21). In p53-depleted human ovarian cancer
(22) and epithelial ovarian
cancer with p53 point mutation (23), the expression of miR-34b was shown
to be downregulated, suggesting that miR-34b can inhibit the
proliferation, adhesion and growth of cancer cells. In endometrial
serous adenocarcinoma, the CpG island of the miR-34b promoter
region is hypermethylated, which inhibits the expression of miR-34b
(24). This phenomenon suggested
that miR-34b is able to inhibit the invasion, growth and migration
of cancer cells.
While miR-34b has been found to function as a tumor
suppressor gene in a variety of solid tumor types (15,17–20),
its role in children with acute lymphoblastic leukemia (ALL) has
not been reported, to the best of our knowledge. Abnormalities of
11q23, which contains the miR-34b gene (25), are the most common chromosomal
variations in certain hematopoietic malignancies, occurring in
60–70% of children with ALL. Therefore, it is of particular
interest to study the roles of miR-34b in regulating the
proliferation of leukemic cells and in the pathogenesis of
pediatric leukemia. The present study used reverse-transcription
quantitative polymerase chain reaction (qRT-PCR) and
methylation-specific PCR (MSP) to examine the expression levels and
CpG island methylation status of the miR-34b gene promoter in
pediatric ALL and analyze its clinical significance. Furthermore,
leukemia cells were treated with 5-aza-2-deoxycytidine (5-aza-2-dC)
to examine the effects of miR-34b promoter methylation in leukemia.
Furthermore, K562 cells were transfected with miR-34b mimics to
evaluate its effects on cell proliferation.
Materials and methods
Cell lines
The U937 (CRL-2367™), HL-60 (CCL-240™), MV4-11
(CRL-9591™), M2R (ABT-737 resistant MV4-11), K562 (CCL-243™) and
DAMI (CRL-9792™) leukemia cell lines were purchased from the
American Type Culture Collection (Manassas, VA, USA). The CCRF and
Raji leukemia cell lines were a kind gift from Professor Jianrong
Wang at Cyrus Tang Blood Hematology Center of Soochow University
(Soochow, China). All cells were cultured in RPMI 1640 (Hyclone; GE
Healthcare, Little Chalfont, UK) containing 10% fetal bovine serum
(FBS; Hyclone) in a humidified incubator (Midi40; Thermo Fisher
Scientific, Waltham, MA, USA) containing 5% CO2 at
37°C.
Clinical samples
Bone marrow samples were collected from 87 AL
patients at the Blood Center of the Children's Hospital of Soochow
University (Suzhou, China) from December 2010 to January 2013. The
patients' bone marrow mononuclear cells (BMNCs) were used in the
present study, which were isolated using Ficoll solution. Patients
were diagnosed with AL using combined analysis of morphology,
immunology, cytogenetics and molecular biology (MICM) (26). Leukemic fusion genes, including
those for mixed lineage leukemia (MLL), were detected by RT-PCR. A
total of 38 male and 17 female patients with a median age of 5
years (range, 0.1–13.6 years) and a median white blood cell (WBC)
count of 50.74×109/l (range, 2.1–638×109/l)
were diagnosed with ALL. Furthermore, 17 male and 15 female
patients were diagnosed with acute myeloid leukemia (AML) and had a
median age of 6.55 years (range, 0.1–13 years) and a median WBC
count of 43.78×109/l (range,
1.8–598.9×109/l). In addition, normal bone marrow
samples of 29 males and 14 females were collected from the Surgical
Department of the Children's Hospital of Soochow University
(Suzhou, China) as controls. The median age was 6 years (range,
0.1–16 years) and the median WBC was 7.84×109/l
(2.92–18.63×109/l). Treatments for AL included
chemotherapy and extramedullary leukemia prevention. Patients with
ALL were given chemotherapy according to the Children's Cancer
& Leukaemia Group-2008 regimen (27) and AML patients were treated using
state-of-the-art generic chemotherapy. The present study was
approved by the Ethics Committee of the Children's Hospital of
Soochow University (Suzhou, China) and signed informed consent was
provided by the patient's parents or guardians. The prednisone
sensitivity test was performed according to the Children's Cancer
& Leukemia Group 2008 regimen (27).
5-Aza-2′-deoxycytidine (5-aza-2-dC)
treatment
K562 or HL-60 cells were seeded into a six-well
plate (1.0–1.5×106 cells per well; four well per cell
line). 2 µl 5-aza-2-dC (FINC Chemical Technology, Shanghai,
China) was added into two wells of each cell type. Following
incubation for 48 h, 1 ml TRIzol (Thermo Fisher Scientific) was
added to each well. DNA and RNA were extracted from each group for
RT-qPCR and MSP experiments.
RT-qPCR
Total RNA was extracted from monocytes with TRIzol
according to the manufacturer's instructions. 2 µg RNA was
used to generate the cDNA library for amplifying target genes. A
TaqMan MicroRNA Reverse Transcription kit (Thermo Fisher
Scientific) was used to synthesize the cDNA. The reaction mixture
contained 0.15 µl 100 mM deoxynucleotide triphosphate, 1
µl MultiScribe reverse transcriptase, 1.5 µl reverse
transcription buffer (10X), 0.19 µl RNase inhibitor, 4.16
µl nuclease-free water, 3 µl primer and 5 µl
RNA. The following conditions were used for reverse transcription:
16°C for 30 min, 42°C for 30 min, 80°C for 5 min and 4°C for 1
min.
The TaqMan MicroRNA Assay and TaqMan Universal PCR
Master Mix (Thermo Fisher Scientific) were used to amplify the cDNA
in a LightCycler 480® II (Roche, Basel, Switzerland).
The reaction contained 1 µl TaqMan MicroRNA Assay mixture
(20X), 1.3 µl cDNA product mixture, 10 µl TaqMan
Universal PCR Master Mix (2X) and 7.7 µl nuclease-free
water. FAM™ dye (GenePharma, Shanghai, China) was added as the
fluorescence probe. PCR was performed using the following cycling
conditions with incorporation of FAM into the nucleotides: 95°C for
10 min, followed by 55 cycles of 95°C for 15 sec and 60°C for 60
sec. U6 small nuclear (sn)RNA
(5′-GTGCTCGCTTCGGCAGCACATATACTAAAATTGGAACGATACAGAGAAGATTAGCATGGCCCCTGCGCAAGGATGACACGCAAATTCGTGAAGCGTTCCATATTTT-3′)
was used as an internal control to normalize the relative
repression levels of miR-34b mimics
(5′-UAGGCAGUGUCAUUAGCUGAUUG-3′). Melting curve
analysis was performed and the R-value was calculated from the
difference between the target gene in the experimental group
compared to that of the control group, using the 2−ΔΔCt
method where Ct was the cycle threshold (i.e., the cycle number at
which the fluorescence reached the set threshold). ΔCt was
calculated by subtracting the Ct value of the U6 snRNA reference
from the Ct value of miR-34b mimics: ΔCtmiR-34b =
CtmiR-34b − CtU6 snRNA. ΔΔCt was then
calculated by subtracting the ΔCt of the respective sample from the
ΔCt of the control group: ΔΔCt = ΔCtSample −
ΔCtControl. Triplicate experiments were performed for
each sample.
The primers used for PCR amplification were as
follows: miR-34b forward,
5′-TGGTTTAGTTATGTGTGTTGTGT-3′ and reverse,
5′-CAACTACAACTCCCAAACAATCC-3′ (Invitrogen; Thermo
Fisher Scientific).
Genomic DNA isolation and MSP
The TIANamp Genomic DNA kit (Tiangen, Beijing,
China) was used to extract the genomic DNA from cell lines and
monocytes according to the manufacturer's instructions. Briefly,
cells were centrifuged at 400 × g for 5 min. 200 µl GA
buffer was added after removing the supernatant. After proteinase K
treatment, 200 µl GB buffer was added followed by incubation
at 70°C for 10 min. 200 µl ethanol was then added and DNA
was purified using the column included in the kit. The DNA
concentration was measured with a BioMate™ 35 ultraviolet
spectrophotometer (Chemlab Corp., Shanghai, China). The EZ
Methylation-Gold kit (Zymo Research, Irvine, CA, USA) was used to
modify the genomic DNA with sodium bisulfite according to the
manufacturer's instructions. Conversion Reagent was prepared by
adding 900 µl water, 50 µl M-Dissolving Buffer and
300 µl M-Dilution Buffer into a tube of CT Conversion
Reagent supplied with the kit. 130 µl CT Conversion Reagent
was mixed with 20 µl genomic DNA and reacted for 10 min at
98°C, followed by 2.5 h at 64°C. 600 µl M-Binding Buffer was
mixed with DNA sample in a Zymo-Spin IC Column (Zymo Research).
After centrifugation at 400 × g for 10 sec, 200 µl
M-Desulphonation Buffer was added onto the same column following
incubation for 15–20 min at room temperature. The column was then
washed twice with M-Wash buffer. 10–20 µl M-Elution Buffer
was used to elute the modified genomic DNA. Takara Taq™ (Takara
Bio, Inc., Otsu, Japan) was used for methylation-specific PCR. The
sequences of the primers were as follows (12): Methylated miR-34b forward,
5′-TTTAGTTACGCGTGTTGTGC-3′ and reverse,
5′-ACTACAACTCCCGAACGATC-3′; unmethylated miR-34b
forward, 5′-TGGTTTAGTTATGTGTGTTGTGT-3′ and reverse,
5′-CAACTACAACTCCCAAACAATCC-3′.
Cell transfection
Transfection was performed using Lipofectamine 2000
(Thermo Fisher Scientific) according to the manufacturer's
instructions. In brief, 4×105 K562 cells were cultured
in 2 ml antibiotic-free RPMI 1640 medium containing 10% FBS in a
six-well plate one day prior to transfection. 24 µl Homo
sapiens (hsa)-miR34b mimics and 24 µl negative control
(GenePharma) were mixed with 226 µl Opti-MEM (Invitrogen),
respectively. 12 µl Lipofectamine 2000 was mixed with 238
µl Opti-MEM and then mixed with the hsa-miR-34b mimics- or
negative control-Opti-MEM solution. After incubation for 20 min at
room temperature, the mixtures were added drop-wise to the cultured
cells. Culture medium was replaced 4–6 h after transfection. Each
transfection was performed in three replicates. The sequences of
the FAM-labeled miRNAs were as follows: hsa-miR-34b mimics sense,
5′-CAAUCACUAACUCCACUGCCAU-3′ and anti-sense,
5′-GGCAGUGGAGUUAGUGAUUGUU-3′; negative control sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′.
Flow cytometry
Cells were harvested 48 h after transfection.
Following two washes with phosphate-buffered saline, cells were
re-suspended in 600 µl phosphate-buffered saline and
analyzed on a FACScan flow cytometer (BD Biosciences, San Jose, CA,
USA). Data were analyzed using CellQuest Pro 5.2 software (BD
Biosciences).
Cell Counting Kit-8 (CCK-8) proliferation
assay
After transfection, cells in the exponential phase
were collected in RPMI 1640 medium containing 10% FBS. Cell
suspension (200 µl) was added into each well of five 96-well
plates at a concentration of 2.5×104/ml. Cells were
analyzed at the time-points of 24, 48, 72, 96 and 120 h. Five
replicates of the blank control, negative control, experimental
group and culture medium control were assessed at each time-point.
20 µl CCK-8 (Shanghai Yes Service Biotech, Shanghai, China)
was added into each well and cells were incubated for another 2 h.
The optical density at 450 nm (OD450) was then assessed
using a microplate reader (MultiSkan FC, Thermo Fisher Scientific)
and regarded as a measure of the number of viable cells.
Statistical analysis
All statistical analyses were performed using SPSS
version 17.0 software (SPSS, Inc., Chicago, IL, USA). Values are
expressed as the mean ± standard deviation. Student's t-test was
used for comparisons among multiple groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
miR-34b is downregulated in leukemia
cells
First, RT-qPCR analysis was performed to compare the
expression levels of miR-34b between 8 leukemia cell lines as well
as the BMNCs of 42 ALL patients, 20 AML patients, 11 patients with
MLL and 20 age-matched normal individuals. Compared with that in
normal controls (5.22±1.15), the relative expression of miR-34b in
leukemia cell lines (0.03±0.03; P<0.01) as well as in BMNCs
cells of patients with ALL (1.65±0.69; P<0.05), AML (0.18±0.06;
P<0.01) and MLL (0.64±0.34; P<0.01) was significantly
downregulated (Table I, Fig. 1). This result indicated that
miR-34b may represent a diagnostic indicator for leukemia. To
further evaluate the potential of using miR-34b as a clinical
marker for AL, miR-34 expression in AL patients was compared with
their clinical parameters. However, no correlation between the
expression levels of miR-34 and the patients' gender, age, WBC
number, immunophenotype, karyotype, gene fusion, MLL gene
rearrangement or LDH levels were identified (P>0.05) (Table II). Of note, the expression levels
of miR-34b in patients sensitive to the ALL prednisone reaction
(0.67±0.22) were significantly lower than those in insensitive
patients (4.40±2.45; P=0.015) (Table
II).
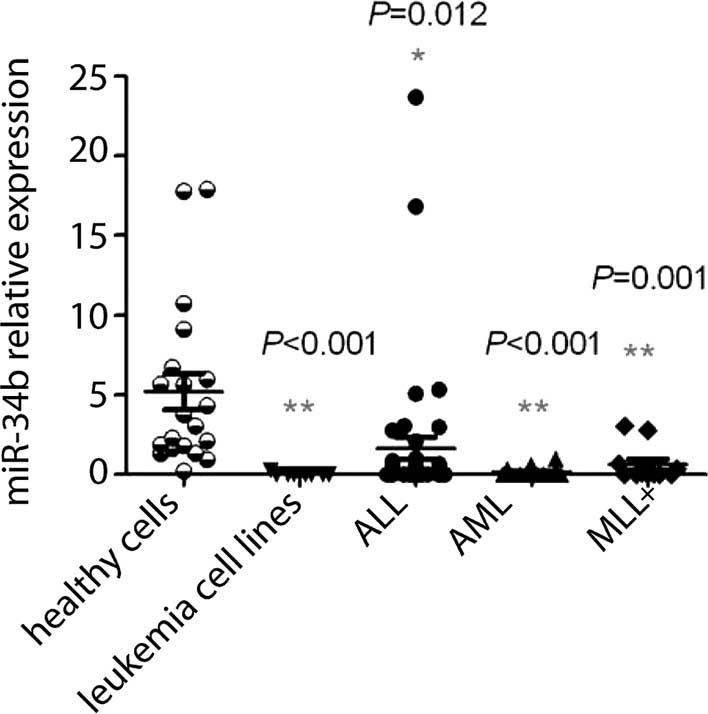 | Figure 1Expression of miR-34b in leukemia
cells. Histogram showing the relative levels of miR-34b in healthy
cells, leukemia cell lines, ALL cells, AML cells and MLL+ cells.
Compared with normal cells, the levels of miR-34b in leukemia cell
lines, ALL cells, AML cells, and MLL+ cells were significantly
lower (P<0.05). MLL+ indicates AL patients with MLL
rearrangement. Leukemia cell lines comprised U937, HL-60, MV4-11,
M2R, K562, Raji, CCRF and DAMI. Each data point represents the
result for one subject/cell line, horizontal bars represent the
mean value and bars indicate the standard deviation.
*P<0.05; **P<0.01 vs. healthy cells.
miR, microRNA; ALL, acute lymphoblastic leukemia; AML, acute
myeloid leukemia; MLL, mixed lineage leukemia. |
 | Table IRelative expression of miR-34b in
different groups. |
Table I
Relative expression of miR-34b in
different groups.
| Group | Cases (n) | Relative
expression | P-value |
|---|
| Normal | 20 | 5.22±1.15 | – |
| Cell lines | 8 | 0.03±0.03 | <0.001 |
| ALL | 42 | 1.65±0.69 | 0.012 |
| AML | 20 | 0.18±0.06 | <0.001 |
| MLL+ | 11 | 0.64±0.34 | 0.001 |
| Table IIAssociation between miR-34b
expression levels and clinical parameters of patients newly
diagnosed with AL. |
Table II
Association between miR-34b
expression levels and clinical parameters of patients newly
diagnosed with AL.
| Clinical
parameter | Number | Relative miR-34b
expression level | P-value |
|---|
| Gender | | | 0.684 |
| Male | 36 | 1.01±0.49 | |
| Female | 26 | 1.41±0.92 | |
| Age (years) | | | 0.797 |
| <1 | 7 | 0.50±0.43 | |
| 1–10 | 49 | 1.34±0.59 | |
| >10 | 6 | 0.60±0.32 | |
| WBC count
(109/l) | | | 0.166 |
| <50 | 28 | 2.16±1.02 | |
| 50–100 | 9 | 0.69±0.28 | |
| >100 | 25 | 0.48±1.75 | |
| Immunosubtype | | | 0.115 |
| Lymphoid | 42 | 1.65±0.69 | |
| Myeloid | 20 | 0.18±0.06 | |
| Karyotype | | | 0.740 |
| Normal | 29 | 1.35±0.82 | |
| Abnormal | 33 | 1.03±0.53 | |
| Gene fusion | | | 0.209 |
| Undetectable | 26 | 1.88±1.10 | |
| Abnormal | 36 | 0.67±0.20 | |
| MLL gene
rearrangement | | | 0.603 |
| Negative | 51 | 1.29±0.57 | |
| Positive | 11 | 0.64±0.34 | |
| LDH levels | | | 0.265 |
| <500 U/l | 17 | 2.04±1.39 | |
| ≥500 U/l | 45 | 0.85±0.39 | |
| ALL prednisone
reaction | | | 0.015 |
| Sensitive | 31 | 0.67±0.22 | |
| Insensitive | 11 | 4.40±2.45 | |
miR-34b inhibits leukemia-cell
proliferation
To assess the effects of miR-34b on cell
proliferation, hsa-miR-34b mimics and negative control RNA were
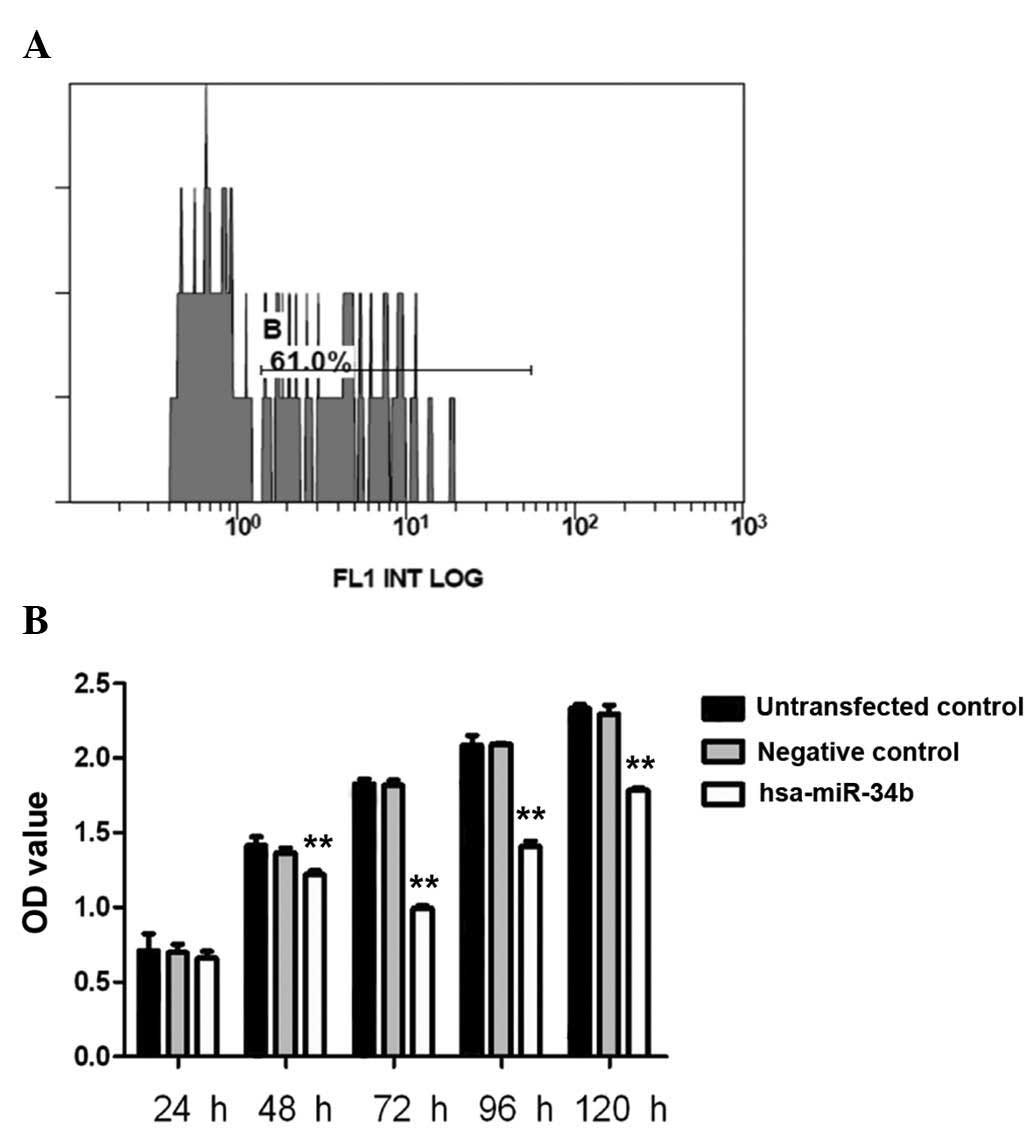
transfected into K562 cells. The transfection efficiency of
hsa-miR-34b in K562 cells was 61%, as determined by flow cytometry
(Fig. 2A). K562-cell proliferation
was then evaluated using the CCK-8 cell proliferation assay. The
OD450-values of hsa-miR-34b-transfected cells were
significantly lower than those of non-transfected cells or negative
control-transfected cells at 48 h, 72 h, 96 h and 120 h (P<0.01)
(Fig. 2B). At 72 h, cell
proliferation was inhibited by 45.7% in hsa-miR-34b
mimic-transfected cells as compared with that in negative control
cells (Fig. 2B).
miR-34b expression is regulated via
methylation of CpG islands in its gene promoter
To examine whether the methylation of CpG island in
the promoter of miR-34b is involved in the regulation of its
expression, MSP analysis was performed to detect the methylation
status of CpG islands in leukemia cell lines as well as in BMNCs
from ALL patients, AML patients MLL patients and healthy controls.
No methylation was detected in cells from all 23 normal control
subjects (results from 13 controls are shown in Fig. 3A). However, methylation was present
in the miR-34b gene promoters of all leukemia cell lines (U937,
HL-60, MV4-11, M2R, K562, Raji, CCRF and DAMI) (Fig. 3B). Among the 31 ALL patients,
methylation was detected in 24 patients (results from 15 patients
are shown in Fig. 3C). Among the
19 AML patients, methylation was detected in eight patients
(results from nine patients are shown in Fig. 3D). The methylation status was then
assessed in all acute leukemia patients. Similarly, no correlation
was found between methylation and patients' gender, age, karyotype,
gene fusion, MLL gene rearrangement, TEL/AML1 gene, Hb number, WBC
count, platelet count or LDH levels (P>0.05) (Table III). However, a significant
difference in miR-34b promoter methylation was detected between
patients with ALL and those with AML (P=0.012) (Table III). Thus, the methylation status
of the miR-34b promoter may be utilized as a marker for the
diagnosis of leukemia sub-types.
 | Figure 3miR-34b is exclusively methylated in
leukemia cells. An MSP assay revealed that (A) miR-34b was
unmethylated in normal cells, (B) miR-34b was methylated in eight
leukemia cell lines comprising U937, HL-60, MV4-11, M2R, K562,
Raji, CCRF and DAMI. (C) miR-34b was methylated in the majority (24
out of 31) of the acute lymphoblastic leukemia cell samples
(results from 15 patients are shown). (D) miR-34b was methylated in
several acute myeloid leukemia cell samples (8 out of 19; results
for nine patients are shown). miR, microRNA; M, methylated; U,
unmethylated; MSP, A methylation-specific polymerase chain
reaction. |
| Table IIIAssociation between miR-34b
methylation and clinical parameters of patients newly diagnosed
with acute leukemia. |
Table III
Association between miR-34b
methylation and clinical parameters of patients newly diagnosed
with acute leukemia.
| Clinical
parameter | miR-34b methylation
status
| P-value |
|---|
| Methylated
(n=32) | Non-methylated
(n=18) |
|---|
| Gender | | | 0.631 |
| Male | 21 (65.6%) | 13 (72.2%) | |
| Female | 11 (34.4%) | 5 (27.8%) | |
| Immunosubtype | | | 0.012 |
| Lymphoid | 24 (75.0%) | 7 (38.9%) | |
| Myeloid | 8 (25%) | 11 (61.1%) | |
| Karyotype | | | 0.585 |
| Normal | 15 (46.9%) | 7 (38.9%) | |
| Abnormal | 17 (53.1%) | 11 (61.1%) | |
| Gene fusion | | | 0.423 |
| Undetectable | 14 (43.8%) | 10 (55.6%) | |
| Abnormal | 18 (56.2%) | 8 (44.4%) | |
| MLL gene
rearrangement | | | 0.292 |
| Positive | 3 (9.4%) | 0 (0%) | |
| Negative | 29 (90.6%) | 18 (100%) | |
| TEL/AML1 gene | | | 0.402 |
| Positive | 3 (9.4%) | 4 (22.2%) | |
| Negative | 29 (90.6%) | 14 (77.8%) | |
| Age (years) | 6.26±3.29 | 6.67±3.47 | 0.685 |
| Hemoglobin
(g/l) | 76.81±25.30 | 80.61±21.86 | 0.596 |
| WBC count
(109/l) | 113.40±163.57 | 92.15±162.15 | 0.660 |
| Platelet count
(109/l) | 46.22±39.45 | 47.67±33.73 | 0.896 |
| LDH levels
(U/l) |
2038.84±3921.65 |
1517.45±1771.48 | 0.597 |
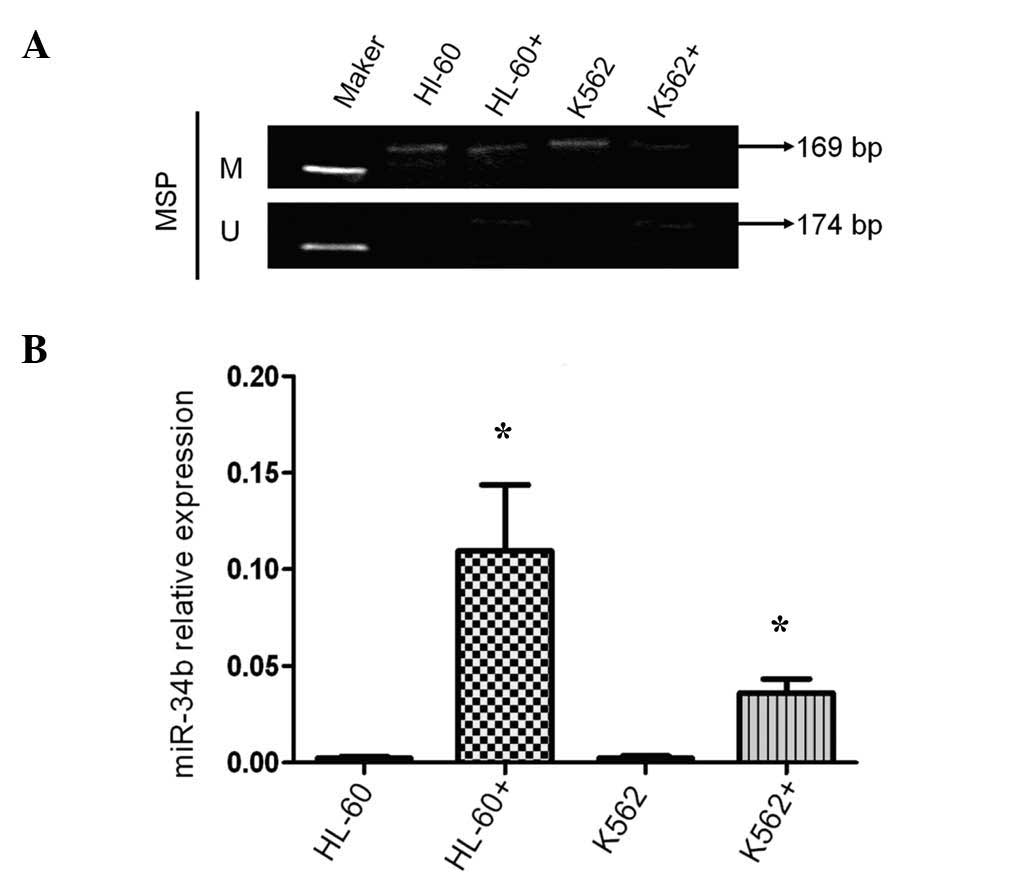
5-Aza-2-dC treatment increases the
expression of miR-34b and decreases the methylation of its
promoter
To evaluate the association between the expression
levels and the CpG island methylation status of the miR-34b gene
promoter, HL-60 and K562 cells were treated with the demethylating
agent 5-aza-2-dC. Methylation of the promoter of miR-34b was
obviously decreased by 5-aza-2-dC (Fig. 4A). Furthermore, miR-34b expression
in 5-aza-2-dC-treated HL-60 and K562 cells was 49.5- and 18.8-fold
increased, respectively, compared with that in untreated cells
(Fig. 4B).
Discussion
In order to study the function of miR-34b and the
methylation of its promoter in AL, the expression levels of miR-34b
were assessed in a panel of leukemia cell lines as well as in
leukemia cells from young patients with various types of AL and
healthy control subjects using RT-qPCR. Compared with that in
normal control subjects, the expression of miR-34b in leukemia cell
lines and leukemia cells of patients with ALL, AML or MLL was
significantly decreased, which is consistent with the findings of
previous studies (28,29). Therefore, it was indicated that
miR-34b is a tumor suppressor gene, which has a role in the
oncogenesis and prognosis of pediatric AL, particularly in MLL. The
MLL gene is located on chromosome 11q23 in the same locus that
encodes miR-34b. The 11q23/MLL rearrangement has been identified as
a specific characteristic of leukemia due to its association with
the leukemia type and poor prognosis; furthermore, the selection of
an individualized treatment strategy is dependent on the presence
of this rearrangement (30). The
World Health Organization has classified the 11q23 rearrangement
separately as '11q23/MLL leukemia' (31). Munoz et al (30) reported that patients with MLL-gene
rearrangement were insensitive to conventional chemotherapy, but
responded to high-dose chemotherapy or stem-cell transplantation.
Thus, detection of miR-34b may assist in the selection of a
patient's chemotherapy regimen.
The present study also analyzed the association
between miR-34b expression levels and clinical characteristics of
patients diagnosed with AL. No significant difference between the
relative expression of miR-34b and gender, age, initial WBC count,
immunophenotype, chromosome fusion, MLL gene rearrangements, LDH
levels at diagnosis or other indicators were identified
(P>0.05). Of note, the relative expression levels of MiR-34b in
MLL patients were decreased compared with those in patients with
non-MLL leukemia; however, this difference was not statistically
significant, presumably due to the limited number of specimens. The
prednisone test, which reflects the early treatment response, is
able to predict the prognosis of ALL (32). In the present study, the relative
expression of miR-34b was significantly different between ALL
patients who were sensitive to the prednisone test and
prednisone-insensitive patients (P<0.05), indicating that the
relative expression of miR-34b may affect the early treatment
response and may also serve as an indicator of poor prognosis newly
diagnosed ALL patients.
Abnormalities in DNA methylation and aberrant
expression of miRNAs were found to have an important role in the
occurrence, development and prognosis of leukemia (33). The CpG island of the miR-34b
promoter was found to be hypermethylated and miR-34b expression was
downregulated in a variety of solid tumor types (15,17–20)
as well as in hematological malignancies (28,29,33,34).
Leucci et al (34) reported
that the CpG island of the miR-34b promoter was hypermethylated and
that miR-34b was silenced in Burkitt's lymphoma without MYC
translocation. Pigazzi et al (28) reported that the miR-34b promoter in
leukemia cell lines was also hypermethylated. Recently, Pigazzi
et al (29) found that
miR-34b expression was decreased in patients newly diagnosed with
AML, and that the miR-34b promoter was hypermethylated in 66% of
AML patients. However, to the best of our knowledge, methylation of
the miR-34b promoter in patients newly diagnosed with ALL has not
been reported. The present study used MSP to detect miR-34b
promoter methylation in patients newly diagnosed with AL and found
that methylation was present in all of the eight leukemia cell
lines assessed (U937, HL-60, MV4-11, M2R, K562, Raji, CCRF and
DAMI), in 24 out of 31 patients (77.42%) newly diagnosed with ALL,
and in 8 out of 19 patients (42.11%) newly diagnosed with AML.
However, no hypermethylation was detected in the 23 normal control
subjects, suggesting that methylation of the miR-34b promoter is
closely associated with hematopoietic malignances. The present
study also found that the percentage of miR-34b promoter
hypermethylation in patients newly diagnosed with ALL was
significantly higher than that in patients newly diagnosed with
AML. Pigazzi et al (29)
reported that the miR-34b promoter was hypermethylated in 66% of
AML patients, which is higher than the ratio determined in the
present study. This difference may be due to the differences in
ethnic groups or the small cohort size.
Pigazzi et al (29) reported that the miR-34b promoter
methylation status in patients newly diagnosed with AML was not
correlated with their clinical parameters, but was associated with
poor prognosis and a lower overall survival. Consistently, the
results of the present study indicated no significant difference
between methylation status and relevant clinical parameters,
including the patient's gender, age, chromosome fusion, TEL/AML1
gene expression, initial hemoglobin count, WBC count, platelet
count and LDH levels, in patients newly diagnosed with AL
(P>0.05). However, the present study found that the miR-34b
promoter methylation level in lymphoid leukemia was significantly
different from that in myeloid leukemia (P<0.05). As patients
with lymphoid leukemia have a higher remission rate and a longer
survival period than those with myeloid leukemia, it may be deduced
that miR-34b methylation is correlated with prognosis and overall
survival rate.
Methyltransferase inhibitors, such as 5-aza-2dC, can
restore the expression of methylation-silenced genes (35). In this study, 5-aza-2-dC treatment
was shown to inhibit the methylation of the miR-34b promoter and
increase the expression of miR-34b. CpG island hypermethylation of
tumor suppressor gene promoters is a common phenomenon in human
leukemia (36). The present study
confirmed that CpG island hypermethylation reduced the expression
of the tumor suppressor miRNA miR-34b, which was regulated via CpG
island methylation in leukemia cell lines and patient samples.
To further assess the suppressive effects of miR-34b
on leukemia-cell proliferation, hsa-miR-34b mimics were transfected
into the K562 leukemia cell line. A CCK-8 assay revealed that high
expression of miR-34b led to the suppression of cell proliferation,
with 72 h being the most effective time-point leading to a decrease
of K562-cell proliferation by almost 50% compared with that of
non-transfected or control-transfected K562 cells. Pigazzi et
al (28) transfected miR-34b
into HL-60 and K562 cells and also found that cell growth and
proliferation were inhibited, and that the cell populations in the
S-phase and G2/M phase of the cell cycle were significantly
reduced, indicating that miR-34b inhibits leukemia-cell
proliferation by causing cell-cycle arrest. The same group also
inoculated miR-34b-transfected HL-60 and K562 cells into NOD-SCID
interleukin-2 receptor gamma-null mice, which resulted in the
formation of obviously smaller tumors compared with those generated
from empty vector-transfected cells, further confirming the role of
miR-34b as a tumor suppressor gene in vivo (30). The results of the present study are
consistent with these two studies, suggesting that miR-34b also has
a tumor suppressor role in pediatric leukemia.
In conclusion, the results of the present study
suggested that miR-34b promoter methylation is likely to be an
important post-transcriptional regulatory mechanism associated with
childhood leukemia. This finding may aid in the development of
novel diagnostic methods and therapies for childhood leukemia.
Acknowledgments
The present study was supported by the National
'Eleventh Five-Year' Major Science and Technology Funding grant
(no. 2007BAI04B03), the National 'Twelfth Five-Year' Major Science
and Technology Funding grant (no. 2011ZX09302-007-01), the National
Natural Science Foundation of China (no. 81100371), the Suzhou
Science and Technology Development Plan 2013 (no. SYS201352) and
the 2012 Suzhou City 'Science and Education Guardian' Youth Science
and Technology Project (no. KJXW2012021).
References
|
1
|
Wang WP: Pediatrics. 8th edition. People's
Medical Publishing House; Beijing: pp. 376–383. 2008, In
Chinese.
|
|
2
|
Pui CH: Recent research advances in
childhood acute lymphoblastic leukemia. J Formos Med Assoc.
109:777–787. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gill Super HJ: A role for epigenetics in
the formation of chromosome translocations in acute leukemia.
Cancer Genet. 208:230–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mourelatos Z, Dostie J, Paushkin S, Sharma
A, Charroux B, Abel L, Rappsilber J, Mann M and Dreyfuss G: miRNPs:
A novel class of ribonucleoproteins containing numerous microRNAs.
Genes Dev. 16:720–728. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alvarez-Garcia I and Miska EA: MicroRNA
functions in animal development and human disease. Development.
132:4653–4662. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Doench JG, Petersen CP and Sharp PA:
siRNAs can function as miRNAs. Genes Dev. 17:438–442. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
He L and Hannon GJ: MicroRNAs: Small RNAs
with a big role in gene regulation. Nat Rev Genet. 5:522–531. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Calin GA and Croce CM: MicroRNAs and
chromosomal abnormalities in cancer cells. Oncogene. 25:6202–6210.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gong JN, Yu J, Lin HS, et al: The role,
mechanism and potentially therapeutic application of microRNA-29
family in acute myeloid leukemia. Cell Death Differ. 21:100–112.
2014. View Article : Google Scholar
|
|
10
|
Moarii M, Boeva V, Vert JP and Reyal F:
Changes in correlation between promoter methylation and gene
expression in cancer. BMC Genomics. 16:8732015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang YY, Tian WP and Mei M: Interaction
between miR-21 and DNA methylation in different breast cancer
cells. Chin J Appl Physiol. 31:220–224. 2015.In Chinese.
|
|
12
|
Garzia L, Andolfo I, Cusanelli E, Marino
N, Petrosino G, De Martino D, Esposito V, Galeone A, Navas L,
Esposito S, et al: MicroRNA-199b-5p impairs cancer stem cells
through negative regulation of HES1 in medulloblastoma. PloS One.
4:e49982009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee KH, Lotterman C, Karikari C, Omura N,
Feldmann G, Habbe N, Goggins MG, Mendell JT and Maitra A:
Epigenetic silencing of MicroRNA miR-107 regulates cyclin-dependent
kinase 6 expression in pancreatic cancer. Pancreatology. 9:293–301.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Noonan EJ, Place RF, Pookot D, Basak S,
Whitson JM, Hirata H, Giardina C and Dahiya R: miR-449a targets
HDAC-1 and induces growth arrest in prostate cancer. Oncogene.
28:1714–1724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Toyota M, Suzuki H, Sasaki Y, Maruyama R,
Imai K, Shinomura Y and Tokino T: Epigenetic silencing of
microRNA-34b/c and B-cell translocation gene 4 is associated with
CpG island methylation in colorectal cancer. Cancer Res.
68:4123–4132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He X, He L and Hannon GJ: The guardian's
little helper: MicroRNAs in the p53 tumor suppressor network.
Cancer Res. 67:11099–11101. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kalimutho M, Di Cecilia S, Del Vecchio
Blanco G, et al: Epigenetically silenced miR-34b/c as a novel
faecal-based screening marker for colorectal cancer. Br J Cancer.
104:1770–1778. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lujambio A, Calin GA, Villanueva A, Ropero
S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso
MS, Faller WJ, et al: A microRNA DNA methylation signature for
human cancer metastasis. Proc Natl Acad Sci USA. 105:13556–13561.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suzuki H, Yamamoto E, Nojima M, Kai M,
Yamano HO, Yoshikawa K, Kimura T, Kudo T, Harada E, Sugai T, et al:
Methylation-associated silencing of microRNA-34b/c in gastric
cancer and its involvement in an epigenetic field defect.
Carcinogenesis. 31:2066–2073. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tsai KW, Wu CW, Hu LY, Li SC, Liao YL, Lai
CH, Kao HW, Fang WL, Huang KH, Chan WC and Lin WC: Epigenetic
regulation of miR-34b and miR-129 expression in gastric cancer. Int
J Cancer. 129:2600–2610. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu C, Cheng H, Shi S, Cui X, Yang J, Chen
L, Cen P, Cai X, Lu Y, Wu C, et al: MicroRNA-34b inhibits
pancreatic cancer metastasis through repressing Smad3. Curr Mol
Med. 13:467–478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kruse JP and Gu W: Modes of p53
regulation. Cell. 137:609–622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Teodoro JG, Parker AE, Zhu X and Green MR:
p53-mediated inhibition of angiogenesis through up-regulation of a
collagen prolyl hydroxylase. Science. 313:968–971. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Feng Z and Levine AJ: The regulation of
energy metabolism and the IGF-1/mTOR pathways by the p53 protein.
Trends Cell Biol. 20:427–434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raver-Shapira N, Marciano E, Meiri E,
Spector Y, Rosenfeld N, Moskovits N, Bentwich Z and Oren M:
Transcriptional activation of miR-34a contributes to p53-mediated
apoptosis. Mol Cell. 26:731–743. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao R and Zhang R: Combined analysis on
morphology, immunology, cytogenetics and molecular biology (MICM)
classification of 55 patients with acute promyelocytic leukemia. J
Exp Hematol. 12:147–150. 2004.In Chinese.
|
|
27
|
Gao C, Zhao XX, Li WJ, Cui L, Zhao W, Liu
SG, Yue ZX, Jiao Y, Wu MY and Li ZG: Clinical features, early
treatment responses and outcomes of pediatric acute lymphoblastic
leukemia in China with or without specific fusion transcripts: A
single institutional study of 1,004 patients. Am J Hematol.
87:1022–1027. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pigazzi M, Manara E, Baron E and Basso G:
MiR-34b targets cyclic AMP-responsive element binding protein in
acute myeloid leukemia. Cancer Res. 69:2471–2478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pigazzi M, Manara E, Bresolin S, Tregnago
C, Beghin A, Baron E, Giarin E, Cho EC, Masetti R, Rao DS, et al:
MicroRNA-34b promoter hypermethylation induces CREB overexpression
and contributes to myeloid transformation. Haematologica.
98:602–610. 2013. View Article : Google Scholar :
|
|
30
|
Munoz L, Nomdedéu JF, Villamor N, Guardia
R, Colomer D, Ribera JM, Torres JP, Berlanga JJ, Fernández C,
Llorente A, et al: Acute myeloid leukemia with MLL rearrangements:
Clinicobiological features, prognostic impact and value of flow
cytometry in the detection of residual leukemic cells. Leukemia.
17:76–82. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cox MC, Panetta P, Venditti A, Del Poeta
G, Maurillo L, Tamburini A, Del Principe MI and Amadori S:
Fluorescence in situ hybridization and conventional cytogenetics
for the diagnosis of 11q23+/MLL+ translocation in leukaemia. Br J
Haematol. 121:953–955. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Inaba H and Pui CH: Glucocorticoid use in
acute lymphoblastic leukaemia. Lancet Oncol. 11:1096–1106. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang LQ and Chim CS: DNA methylation of
tumor-suppressor miRNA genes in chronic lymphocytic leukemia.
Epigenomics. 7:461–473. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Leucci E, Cocco M, Onnis A, De Falco G,
van Cleef P, Bellan C, van Rijk A, Nyagol J, Byakika B, Lazzi S, et
al: MYC translocation-negative classical Burkitt lymphoma cases: An
alternative pathogenetic mechanism involving miRNA deregulation. J
Pathol. 216:440–450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Baylin SB, Herman JG, Graff JR, Vertino PM
and Issa JP: Alterations in DNA methylation: A fundamental aspect
of neoplasia. Adv Cancer Res. 72:141–196. 1998. View Article : Google Scholar
|
|
36
|
Cahill N and Rosenquist R: Uncovering the
DNA methylome in chronic lymphocytic leukemia. Epigenetics.
8:138–148. 2013. View Article : Google Scholar : PubMed/NCBI
|