Introduction
Adenosine monophosphate (AMP)-activated protein
kinase (AMPK) is widely recognized as a central regulator of
cellular metabolism and energy homeostasis (1,2).
AMPK has been discovered as an enzyme that catalyzes the
phosphorylation of acetyl coenzyme A (CoA) carboxylase, which
regulates lipid synthesis (2).
AMPK activity is upregulated by the elevation of the AMP/adenosine
triphosphate (ATP) ratio in response to various types of
physiological and pathological stress, resulting in restoring
cellular enzyme balance by ATP generating pathways (3). In addition, activated AMPK suppresses
ATP utilizing pathways. Therefore, AMPK is currently known to
regulate metabolic homeostasis throughout the body (2).
Bone metabolism is predominantly regulated by two
types of functional cells, osteoblasts and osteoclasts (4). The former cells are responsible for
bone formation, while the latter cells are responsible for bone
resorption. Constant bone mass is maintained by bone remodeling,
which comprises osteoclastic bone resorption followed by
osteoblastic bone formation. Disruption to bone remodeling can
result in metabolic bone disease, including osteoporosis.
Concerning the association between AMPK and bone metabolism, it has
been demonstrated that AMPK activation stimulates osteoblast
differentiation and bone formation, resulting in increased bone
mass (5). It has been previously
reported that vascular endothelial growth factor synthesis induced
by basic fibroblast growth factor is regulated by AMPK in
osteoblast-like MC3T3-E1 cells (6). However, the precise role of AMPK in
osteoblasts remains to be fully elucidated.
Osteoprotegerin (OPG) is an essential protein
secreted from osteoblasts, which inhibits osteoclast activation and
its differentiation, and a member of the tumor necrosis factor
receptor family along with receptor activator of nuclear factor-κB
(RANK) (7). OPG binds to RANK
ligand (RANKL) as a decoy receptor, and prevents RANKL from binding
to RANK, resulting in the suppression of bone resorption (7). It has been demonstrated that RANKL
knockout mice suffer from severe osteopetrosis (8), suggesting that RANKL is a key
regulator of osteoclastogenesis. The RANK/RANKL/OPG axis is
currently recognized as a major regulatory system for osteoclast
formation and activity (9).
Prostaglandins (PGs) are autocrine/paracrine
modulators in the bone metabolism (10). Among them, PGE2 has been
recognized as an important mediator of bone remodeling (11). It has been previously reported that
PGE2 stimulates OPG synthesis via the activation of p38
mitogen-activated protein (MAP) kinase, p44/p42 MAP kinase and
stress activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK)
in osteoblast-like MC3T3-E1 cells (12). However, the detailed mechanism
underlying PGE2-stimulated OPG synthesis in osteoblasts
remains to be fully elucidated.
In the present study, the involvement of AMPK in the
synthesis of OPG induced by PGE2 in osteoblast-like
MC3T3-E1 cells was investigated. The current study aimed to
investigate whether AMPK positively regulates the
PGE2-stimulated OPG synthesis via the SAPK/JNK pathway
in these cells.
Materials and methods
Materials
PGE2 was obtained from Sigma-Aldrich (St.
Louis, MO, USA). The mouse OPG enzyme-linked immunosorbent assay
(ELISA) kit was purchased from R&D Systems, Inc. (Minneapolis,
MN, USA). Compound C was obtained from Calbiochem; EMD Millipore
(Billerica, MA, USA). Polyclonal rabbit phosphorylated (p)-AMPKα
(Thr-172; cat. no. 2531), p-AMPKβ (Ser-108; cat. no. 4181),
p-acetyl-CoA carboxylase (cat. no. 3661), p38 MAP kinase (cat. no.
9212), p-p44/p42 MAP kinase (cat. no. 9101), p44/p42 MAP kinase
(cat. no. 9102), SAPK/JNK (cat. no. 9252) antibodies, and
monoclonal rabbit p-p38 MAP kinase (cat. no. 4511) and p-SAPK/JNK
(cat. no. 4668) antibodies, were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) antibodies (sc-25778) were obtained from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The Enhanced
Chemiluminescence (ECL) Western Blotting Detection System was
purchased from GE Healthcare Life Sciences (Chalfont, UK).
Acrylamide monomer, Tris(hydroxymethyl)amino-methane, sodium
dodecyl sulfate (SDS), dithiothreitol and glycerol were obtained
from Nacalai Tesque, Inc. (Kyoto, Japan). PGE2 was
dissolved in ethanol. Compound C was dissolved in dimethyl
sulfoxide (Sigma-Aldrich). The maximum concentration of ethanol or
dimethyl sulfoxide was 0.1%, which did not affect either the assay
for OPG or the detection of protein levels using western blot
analysis.
Cell culture
Cloned osteoblast-like MC3T3-E1 cells, which were
originally derived from newborn mouse calvaria (13), were maintained as described
previously (14). Briefly, the
cells were cultured in α-minimum essential medium (α-MEM;
Sigma-Aldrich) containing 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in a
humidified atmosphere of 5% CO2/95% air. The cells were
seeded into 35-mm diameter dishes (5×104 cells/dish) or
90-mm diameter dishes (2×105 cells/dish) in α-MEM
containing 10% FBS. Following culture for five days, the medium was
exchanged for α-MEM containing 0.3% FBS. The cells were then used
for experiments after 48 h.
Assay for OPG
The cultured cells were pretreated with 0.3, 1, 3 or
10 µM compound C for 60 min, then stimulated with 10
µM of PGE2 or vehicle in 1 ml α-MEM containing
0.3% FBS, and were incubated for 48 h at 37°C. The conditioned
medium was collected, and the OPG concentration in the medium was
measured using the mouse OPG ELISA kit according to the
manufacturer's protocol.
Western blot analysis
The cultured cells were pretreated with 1, 3 or 10
µM compound C for 60 min, and then were stimulated with 10
µM PGE2 or vehicle for 1, 3, 5, 10, 20, 30 or 60
min. The cells were then washed twice with phosphate-buffered
saline (Sigma-Aldrich) and then lysed, homogenized and sonicated
900 µl lysis buffer containing 62.5 mM Tris/HCl, pH 6.8, 2%
SDS, 50 mM dithiothreitol and 10% glycerol. SDS-polyacrylamide gel
electrophoresis was performed by the method described by Laemmli
(15) in 10% polyacrylamide gels.
The protein was fractionated and transferred onto an Immun-Blot
polyvinylidene difluoride (PVDF) membrane (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Western blot analysis was performed as
described previously (16) using
p-AMPKα (Thr-172), p-AMPKβ (Ser-108), p-acetyl-CoA carboxylase,
p-p38 MAP kinase, p38 MAP kinase, p-p44/p42 MAP kinase, p44/p42 MAP
kinase, p-SAPK/JNK, SAPK/JNK and GAPDH antibodies as the primary
antibodies at a dilution of 1:1,000 in 5% milk in Tris-buffered
saline (20 mM Tris/HCl, pH 7.6, 137 mM NaCl) with 0.1% Tween-20
(TBST) overnight at 4°C. Goat-anti rabbit IgG horseradish
peroxidase-labeled antibodies (074-1506; KPL, Inc., Gaithersburg,
MD, USA) were used as the secondary antibodies at a dilution of
1:1.000 in 5% milk in TBST for 1 h at room temperature. Peroxidase
activity on the PVDF membrane was visualized on X-ray film (Super
RX; Fujifilm, Tokyo, Japan) by means of the ECL Western Blotting
Detection System.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The cultured cells were pretreated with 10 µM
compound C or vehicle for 60 min, then were stimulated by 10
µM PGE2 or vehicle in α-MEM containing 0.3% FBS
for 3 h. Total RNA was isolated and reverse transcribed into
complementary DNA using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and the Omniscript Reverse Transcriptase kit
(Qiagen Inc., Valencia, CA, USA), respectively. RT-qPCR was
performed using a LightCycler system (version 2.0; Roche
Diagnostics, Basel, Switzerland) in capillaries with the FastStart
DNA Master SYBR Green I provided with the RightCycler FastStart DNA
kit (Roche Diagnostics). Sense and antisense primers for mouse OPG
or GAPDH mRNA were purchased from Takara Bio, Inc. (Otsu, Japan;
primer set ID, MA026526). Amplification of the correct PCR products
was confirmed by melting curve analysis according to the
manufacturer's instructions. The OPG mRNA levels were normalized to
those of GAPDH mRNA.
Densitometric analysis
A densitometric analysis of the protein expression
was performed using a scanner (GT-F600; Seiko Epson Corporation,
Nagano, Japan) and an ImageJ analysis software, version 1.48
(National Institutes of Health, Bethesda, MD, USA). The
background-subtracted signal intensity of each phosphorylation
signal was normalized to the respective total protein signal and
plotted as the fold increase in comparison to control cells without
stimulation.
Statistical analysis
The data were analyzed by an analysis of variance,
followed by the Bonferroni method for multiple comparisons between
pairs. P<0.05 was considered to indicate a statistically
significant difference. All data are presented as the mean ±
standard error of triplicate determinations from three independent
cell preparations.
Results
Effects of PGE2 on the
phosphorylation of AMPK or acetyl-CoA carboxylase in MC3T3-E1
cells
It has been previously established that the
phosphorylation of AMPK is essential for its activation (17). Therefore, in order to clarify
whether AMPK is activated by PGE2 in osteoblast-like
MC3T3-E1 cells, the effect of PGE2 on the
phosphorylation of AMPK was investigated by western blot analysis.
PGE2 was observed to significantly induce the
phosphorylation of AMPKα (Thr-172) and AMPKβ (Ser-108). The effects
of PGE2 on the phosphory-lation of AMPK α and AMPKβ
reached their peak 1 min and 5 min subsequent to the stimulation,
respectively, and reduced thereafter (Fig. 1). It is widely accepted that AMPK
induces the phosphorylation of acetyl-CoA carboxylase as a direct
substrate of AMPK, and regulates it (2). Thus, the effect of PGE2 on
the phosphorylation of acetyl-CoA carboxylase was investigated in
MC3T3-E1 cells. PGE2 significantly induced the
phosphorylation of acetyl-CoA carboxylase, and the effect on the
phosphorylation reached its peak 5 min subsequent to stimulation
(Fig. 1).
Effect of compound C on the
PGE2-stimulated OPG release in MC3T3-E1 cells
It has been previously reported that PGE2
stimulates OPG synthesis via the activation of p38 MAP kinase,
p44/p42 MAP kinase and SAPK/JNK in osteoblast-like MC3T3-E1 cells
(12). In order to elucidate
whether AMPK serves a role in the PGE2-induced synthesis
of OPG in MC3T3-E1 cells, the effect of compound C, an inhibitor of
AMPK (18), on the release of OPG
induced by PGE2 was investigated. Compound C, which by
itself had minimal effect on the levels of OPG, significantly
reduced the PGE2-stimulated OPG release in a
dose-dependent manner in the range between 0.3 and 10 µM
(Fig. 2). A 10 µM dose of
compound C resulted in an approximately 40% inhibition in the
PGE2-effect.
Effect of compound C on the
PGE2-stimulated phosphorylation of acetyl-CoA
carboxylase in MC3T3-E1 cells
In order to investigate whether compound C functions
as an inhibitor of AMPK in osteoblast-like MC3T3-E1 cells, the
effect of compound C on the PGE2-induced phosphorylation
of acetyl-CoA carboxylase was examined. Compound C significantly
suppressed the PGE2-induced phosphorylation of
acetyl-CoA carboxylase (Fig.
3).
Effect of compound C on the
PGE2-induced OPG mRNA expression in MC3T3-E1 cells
In order to elucidate whether the suppression of the
PGE2-stimulated OPG synthesis by compound C is mediated
via transcriptional events in MC3T3-E1 cells, the effect of
compound C on the PGE2-induced expression levels of OPG
mRNA were investigated. Compound C was observed to significantly
reduce the PGE2-induced OPG mRNA expression (Fig. 4).
Effects of compound C on the
PGE2-stimulated phosphorylation of p38 MAP kinase,
p44/p42 MAP kinase or SAPK/JNK in MC3T3-E1 cells
Regarding the intracellular signaling of
PGE2 in osteoblasts, it has been previously demonstrated
that PGE2 induces the activation of p38 MAP kinase,
p44/p42 MAP kinase and SAPK/JNK in osteoblast-like MC3T3-E1 cells,
and that these MAP kinases function as positive regulators in the
PGE2-stimulated OPG synthesis in these cells (12). Therefore, the association between
AMPK and these MAP kinases in the PGE2-stimulated OPG
synthesis was investigated in MC3T3-E1 cells. Firstly the effects
of compound C on the PGE2-induced phosphorylation of p38
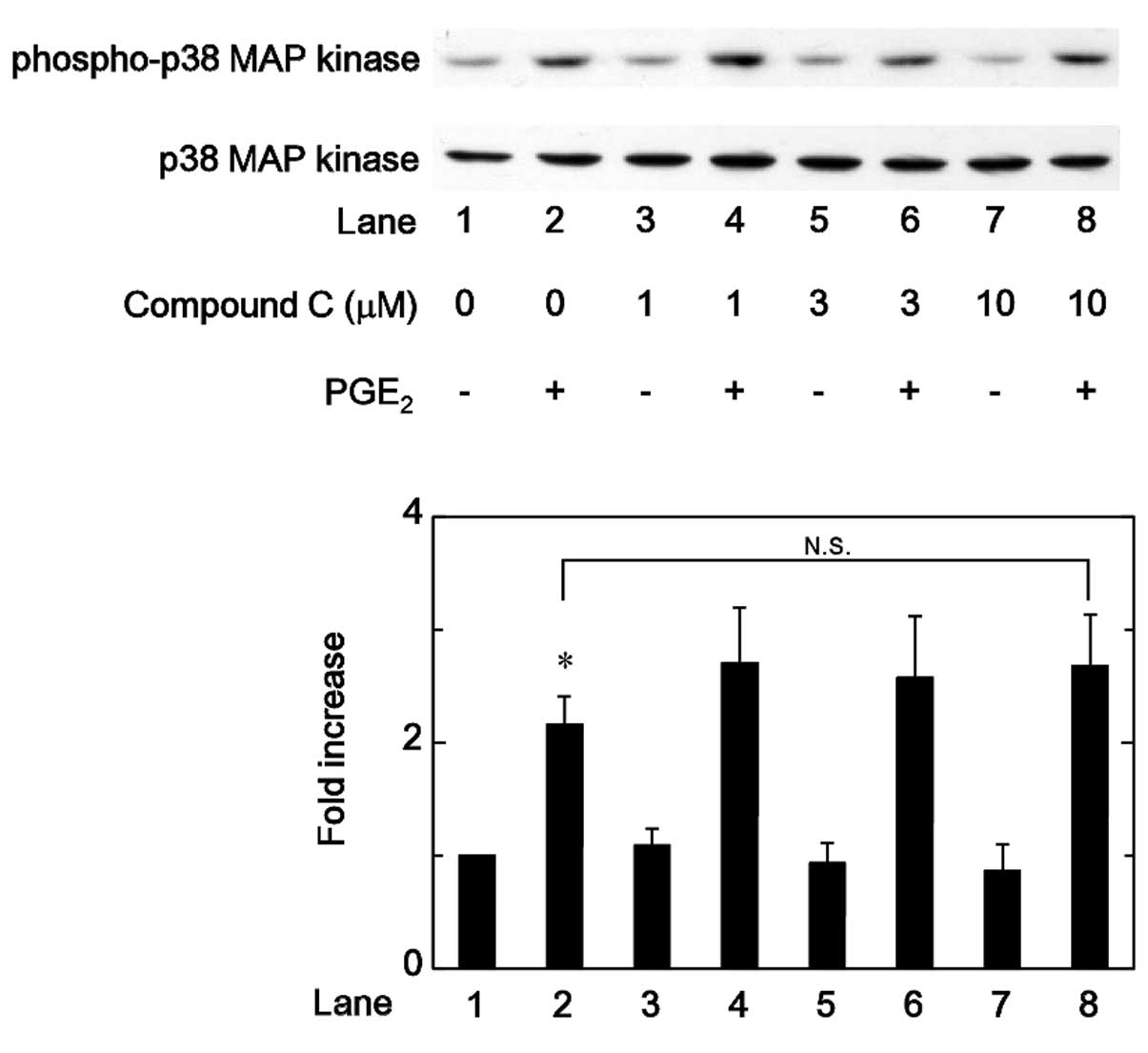
MAP kinase or p44/p42 MAP kinase were examined. However, compound C
was not observed to affect the PGE2-induced
phosphorylation of p38 MAP kinase or p44/p42 MAP kinase in the
range between 1 and 10 µM (Figs. 5 and 6). By contrast, compound C significantly
attenuated the phosphorylation of SAPK/JNK induced by
PGE2 in a dose-dependent manner in the range between 1
and 10 µM (Fig. 7).
Discussion
In the present study, it was demonstrated that the
phosphorylation of AMPKα (Thr-172) and AMPKβ (Ser-108) was markedly
induced by PGE2 in osteoblast-like MC3T3-E1 cells. AMPK
exists as a heterotrimeric complex consisting of three subunits,
which are designated α, β and γ (1). Among the three AMPK subunits, the
α-subunit is recognized as a catalytic site, whereas the β- and
γ-subunits are regulatory sites (17). It is currently recognized that the
phosphorylation of the α-subunit is essential for AMPK activation,
while the phosphorylation of the β-subunit is required for correct
activation of AMPK (17). In the
current study, it was demonstrated that PGE2
significantly stimulated the phosphorylation of acetyl-CoA
carboxylase, a direct substrate of AMPK, in MC3T3-E1 cells. The
time course of PGE2-induced AMPKα phosphorylation was
more rapid than that of acetyl-CoA carboxylase phosphorylation.
Based on these observations, it is suggested that PGE2
induces AMPK activation in osteoblast-like MC3T3-E1 cells.
In a previous study (12), it was reported that PGE2
stimulates OPG synthesis in osteoblast-like MC3T3-E1 cells. In the
present study, the involvement of AMPK in the
PGE2-stimulated OPG synthesis in MC3T3-E1 cells was
investigated, and it was demonstrated that compound C, an inhibitor
of AMPK (18), reduced the release
of OPG induced by PGE2. It was additionally observed
that compound C significantly attenuated the phosphorylation of
acetyl-CoA carboxylase stimulated by PGE2. Therefore,
the observations of the current study suggest that
PGE2-activated AMPK acts as a positive regulator in OPG
release. Furthermore, it was demonstrated that compound C
significantly reduced the PGE2-mRNA expression levels of
OPG. Taking these observations into account, it is suggested that
PGE2 stimulates the synthesis of OPG, at least in part,
via AMPK activation in osteoblast-like MC3T3-E1 cells.
It is widely accepted that the MAP kinase
superfamily serves a central role in a variety of cellular
functions, including proliferation, differentiation and survival
(19). Three major MAP kinases,
p38 MAP kinase, p44/p42 MAP kinase and SAPK/JNK, are recognized as
central elements used by mammalian cells to transduce diverse types
of messages (20). Regarding the
signaling mechanism of the PGE2-stimulated OPG synthesis
in osteoblasts, it has been previously demonstrated that
PGE2 stimulates the activation of p38 MAP kinase,
p44/p42 MAP kinase and SAPK/JNK in osteoblast-like MC3T3-E1 cells,
and that three MAP kinases are implicated in the
PGE2-stimulated OPG synthesis (12). In order to establish how AMPK
functions in PGE2-stimulated OPG synthesis in MC3T3-E1
cells, the association between AMPK and three MAP kinases was
investigated in the current study. It was demonstrated that
compound C suppressed the PGE2-induced phosphorylation
of SAPK/JNK without affecting the phosphorylation of p38 MAP kinase
or p44/p42 MAP kinase. Based on the observations of the present
study, it is suggested that PGE2 induces the activation
of SAPK/JNK via AMPK in osteoblast-like MC3T3-E1 cells. In the
present study, it was demonstrated that the maximum effect of
PGE2 on AMPKα phosphorylation was observed at 1 min
subsequent to stimulation. By contrast, a previously study
demonstrated that PGE2-induced SAPK/JNK phosphorylation
reached its peak at 20 min subsequent to stimulation (12). The time course of the
PGE2-induced AMPK phosphorylation appears to be more
rapid than that of SAPK/JNK phosphorylation, suggesting that the
PGE2-induced activation of SAPK/JNK occurs subsequent to
AMPK activation. Taking the current and previous studies into
account, it is suggested that AMPK acts upstream of SAPK/JNK and
positively regulates the PGE2-stimulated OPG synthesis
in osteoblast-like MC3T3-E1 cells. The potential mechanism of AMPK
in PGE2-induced OPG synthesis in osteoblasts
investigated here is summarized in Fig. 8.
AMPK is generally recognized as a key sensor in
cellular energy homeostasis (1).
Regarding AMPK in osteoblasts, it has been demonstrated that
metformin, an activator of AMPK, increases collagen-1 and
osteocalcin mRNA expression, stimulates alkaline phosphatase
activity and enhances cell mineralization in osteoblast-like
MC3T3-E1 cells (21). In addition,
AMPK activation reportedly inhibits palmitate-induced apoptosis in
osteoblasts (22). Furthermore,
AMPK has been observed to stimulate osteoblast differentiation via
induction of runt-related transcription factor 2 expression
(23). Thus, these observations
lead to the hypothesis that AMPK activation directs osteoblasts
toward stimulating bone formation. PGE2 is a well-known
autocrine/paracrine regulator of osteoblasts and acts as an
important mediator of bone remodeling (10). By contrast, OPG, which prevents the
biological effects of RANKL as a decoy receptor, negatively
regulates RANKL-mediated osteoclastic bone resorption (7). Therefore, the results of the present
study indicate that PGE2-activated AMPK in osteoblasts
functions as a modulator of bone metabolism via OPG synthesis,
resulting in the upregulation of bone formation, and these results
may provide a novel insight into bone metabolism. Further
investigation would be necessary to clarify the exact roles of AMPK
in bone metabolism.
The results of the current study indicate that AMPK
functions as a positive regulator in PGE2-stimulated OPG
synthesis via SAPK/JNK activation in osteoblasts.
Acknowledgments
The authors would like to thank Mrs. Yumiko Kurokawa
for her technical assistance. The current study was supported in
part by the Grant-in-Aid for Scientific Research from the Ministry
of Education, Science, Sports and Culture of Japan (grant no.
19591042) and the Research Funding for Longevity Sciences from the
National Center for Geriatrics and Gerontology, Japan (grant nos.
23-9 and 25-4).
References
|
1
|
Fogarty S and Hardie DG: Development of
protein kinase activators: AMPK as a target in metabolic disorders
and cancer. Biochim Biophys Acta. 1804:581–591. 2010. View Article : Google Scholar
|
|
2
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rutter GA and Leclerc I: The AMP-regulated
kinase family: Enigmatic targets for diabetes therapy. Mol Cell
Endocrinol. 297:41–49. 2009. View Article : Google Scholar
|
|
4
|
Karsenty G and Wagner EF: Reaching a
genetic and molecular understanding of skeletal development. Dev
Cell. 2:389–406. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shah M, Kola B, Bataveljic A, Arnett TR,
Viollet B, Saxon L, Korbonits M and Chenu C: AMP-activated protein
kinase (AMPK) activation regulates in vitro bone formation and bone
mass. Bone. 47:309–319. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kato K, Tokuda H, Adachi S,
Matsushima-Nishiwaki R, Natsume H, Yamakawa K, Gu Y, Otsuka T and
Kozawa O: AMP-activated protein kinase positively regulates
FGF-2-stimulated VEGF synthesis in osteoblasts. Biochem Biophys Res
Commun. 400:123–127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simonet WS, Lacey DL, Dunstan CR, Kelley
M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et
al: Osteoprotegerin: A novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kong YY, Yoshida H, Sarosi I, Tan HL,
Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G,
Itie A, et al: OPGL is a key regulator of osteoclastogenesis,
lymphocyte development and lymph-node organogenesis. Nature.
397:315–323. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Theoleyre S, Wittrant Y, Tat SK, Fortun Y,
Redini F and Heymann D: The molecular triad OPG/RANK/RANKL:
Involvement in the orchestration of pathophysiological bone
remodeling. Cytokine Growth Factor Rev. 15:457–475. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hikiji H, Takato T, Shimizu T and Ishii S:
The roles of prostanoids, leukotrienes and platelet-activating
factor in bone metabolism and disease. Prog Lipid Res. 47:107–126.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu Z, Fu C, Li X, Song Y, Li C, Zou M,
Guan Y and Zhu Y: Prostaglandin E2 promotes endothelial
differentiation from bone marrow-derived cells through AMPK
activation. PloS One. 6:e235542011. View Article : Google Scholar
|
|
12
|
Yamamoto N, Tokuda H, Kuroyanagi G,
Mizutani J, Matsushima-Nishiwaki R, Kondo A, Kozawa O and Otsuka T:
Regulation by resveratrol of prostaglandin E2-stimulated
osteoprotegerin synthesis in osteoblasts. Int J Mol Med.
34:1439–1445. 2014.PubMed/NCBI
|
|
13
|
Sudo H, Kodama HA, Amagai Y, Yamamoto S
and Kasai S: In vitro differentiation and calcification in a new
clonal osteogenic cell line derived from newborn mouse calvaria. J
Cell Biol. 96:191–198. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kozawa O, Tokuda H, Miwa M, Kotoyori J and
Oiso Y: Cross-talk regulation between cyclic AMP production and
phosphoinositide hydrolysis induced by prostaglandin E2
in osteoblast-like cells. Exp Cell Res. 198:130–134. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kato K, Ito H, Hasegawa K, Inaguma Y,
Kozawa O and Asano T: Modulation of the stress-induced synthesis of
hsp27 and alpha B-crystallin by cyclic AMP in C6 rat glioma cells.
J Neurochem. 66:946–950. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hawley SA, Davison M, Woods A, Davies SP,
Beri RK, Carling D and Hardie DG: Characterization of the
AMP-activated protein kinase kinase from rat liver and
identification of threonine 172 as the major site at which it
phosphorylates AMP-activated protein kinase. J Biol Chem.
271:27879–27887. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al: Role of
AMP-activated protein kinase in mechanism of metformin action. J
Clin Invest. 108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
20
|
Widmann C, Gibson S, Jarpe MB and Johnson
GL: Mitogen-activated protein kinase: Conservation of a
three-kinase module from yeast to human. Physiol Rev. 79:143–180.
1999.PubMed/NCBI
|
|
21
|
Kanazawa I, Yamaguchi T, Yano S, Yamauchi
M and Sugimoto T: Metformin enhances the differentiation and
mineralization of osteoblastic MC3T3-E1 cells via AMP kinase
activation as well as eNOS and BMP-2 expression. Biochem Biophys
Res Commun. 375:414–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim JE, Ahn MW, Baek SH, Lee IK, Kim YW,
Kim JY, Dan JM and Park SY: AMPK activator, AICAR, inhibits
palmitate-induced apoptosis in osteoblast. Bone. 43:394–404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jang WG, Kim EJ, Lee KN, Son HJ and Koh
JT: AMP-activated protein kinase (AMPK) positively regulates
osteoblast differentiation via induction of Dlx5-dependent Runx2
expression in MC3T3E1 cells. Biochem Biophys Res Commun.
404:1004–1009. 2011. View Article : Google Scholar
|