Introduction
X-linked adrenal hypoplasia congenita (AHC) is a
rare genetic disorder of adrenal gland development, characterized
by absence or near absence of the permanent zone of the adrenal
cortex (1). Patients with this
condition usually present with primary adrenal failure, including
salt-wasting, hyperpigmentation, failure to thrive, reduced serum
cortisol and aldosterone and increased plasma adrenocorticotropic
hormone (ACTH) (2). The majority
of patients present with symptoms within the first two months of
their life, whereas the remainder present later in childhood
(3,4). The condition is lethal if left
untreated with appropriate steroid hormones (5). Besides adrenal insufficiency,
hypogonadotropic hypogonadism (HHG) is a frequent feature of
X-linked AHC. It is usually recognized during adolescence by the
absence or interruption of normal pubertal development (6,7).
X-linked AHC was originally mapped to Xp21 and the
nuclear receptor sub-family 0, group B, member 1 (NR0B1) was
subsequently identified by positional cloning as the gene
responsible for X-linked AHC and HHG (2,8–11).
The NR0B1 gene consists of two exons and encodes a 470-amino
acid protein termed dosage-sensitive sex-reversal, adrenal
hypoplasia congenital critical region on the X chromosome, protein
1 (DAX1) (12). DAX1 is an orphan
member of the nuclear receptor superfamily. The DAX1
carboxy-terminal domain (CTD) is homologous to the ligand-binding
domain (LBD) of other nuclear receptors, whereas the amino-terminal
domain (NTD), which lacks the typical zinc finger DNA-binding
motif, is composed of three short repeats, each containing an LXXLL
motif (2). DAX1 is predominantly
expressed in the adrenal cortex, gonads, hypothalamus and anterior
pituitary (13,14). Functional studies suggested that
DAX1 is a repressor of gene transcription, acting in part by
inhibiting the activity of another orphan nuclear receptor,
steroidogenic factor 1 (SF-1), encoded by the NR5A1 gene
(15–18). However, the exact role of DAX1 in
the development and function of the adrenal and reproductive axes
has remained elusive.
The present study described the clinical features of
a Chinese male (age, 25 years) with X-linked AHC and HHG. A novel
indel variant of the NR0B1 gene was identified and the
functional effects of the mutation were assessed. The present study
provided insight into the structure-function association of
DAX1.
Materials and methods
Patient
A male patient (age, 25 years) was referred to the
department with the main complaint of absence of pubertal
development. He was full-term at birth, with a normal length and
weight. His current height and weight were 167 cm and 55 kg,
respectively, which were below the 25th percentile of
the Chinese population. Physical examination revealed sparse pubic
hair, small penis (3 cm) and low testicular volume (3 ml
bilaterally) (Tanner stage 1) (19). Mild and diffuse skin pigmentation
was also noticed, with a few hyperpigmented macules on the lips.
Laboratory tests showed low concentrations of serum testosterone
(0.36 ng/ml; normal range, 2.8–8.0 ng/ml) and leutenizing hormone
(1.16 IU/l; normal range, 1.7–8.6 IU/l). The serum
follicle-stimulating hormone concentration was normal (7.16 IU/l;
normal range, 1.5–12.4 IU/l). The patient had a past medical
history of fatigue, nausea, hyperpigmentation and failure to
thrive. The patient was diagnosed with Addison's disease at age 7
on the basis of a high serum ACTH concentration (299 pg/ml; normal
range, up to 46 pg/ml) in the presence of a low serum cortisol
concentration (2.5 ng/ml; normal range, 5–25 ng/ml). The patient
had been treated with hydrocortisone since then. His current serum
ACTH and cortisol concentrations (43.69 pg/ml and 9.4 ng/ml,
respectively) were within the normal range. The patient was born to
non-consanguineous parents, and while no other known case of
X-linked AHC and HHG was present in the family pedigree (Fig. 1), the patient's mother was
identified to be a carrier. The family members with the exception
of the patient were healthy. They did not exhibit any hormone
insufficiency disorders, such as adrenal insufficiency. The ethics
committee of the Shanghai Children's Medical Center (Shanghai,
China) approved the study and written informed consent was obtained
from all subjects.
Molecular analysis of the NR0B1 gene
Peripheral blood samples, to which
ethylenediaminetetraacetic acid (BD, Franklin Lakes, NJ, USA) was
added, were obtained from the patient, the patient's mother and 100
ethnicity-matched healthy individuals. Genomic DNA was isolated
from the peripheral blood leukocytes using the QIAmp DNA Blood kit
(Qiagen, Hilden, Germany). All exons and exon-intron boundaries of
the NR0B1 gene from the proband's genomic DNA were amplified
by polymerase chain reaction (PCR) using the primers listed in
Table I as reported previously
(20). The PCR products were
analyzed by direct DNA sequencing on an ABI 3700 sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA). Only
genomic fragments containing the mutation identified in the proband
were amplified and sequenced for the proband's mother and the
normal controls.
 | Table IPrimers used for amplification and
mutagenesis of NR0B1. |
Table I
Primers used for amplification and
mutagenesis of NR0B1.
| Primer | Sequence
(5′→3′) | Size (bp) |
|---|
|
NR0B1-exon1.1-F |
TCGAACCACCGAGGTCAT | 746 |
|
NR0B1-exon1.1-R |
CTGGTAGCGCCTCTTTACCC | |
|
NR0B1-exon1.2-F |
GCTTGCTCACTAGCTCAAAGC | 859 |
|
NR0B1-exon1.2-R |
TCACGATTTCTTCACCTTTGC | |
|
NR0B1-exon2-F |
TCTTGGACACGTTGCTTCTG | 717 |
|
NR0B1-exon2-R |
GCAGGTTCCATGAAATTGCT | |
|
195_207delinsTG-F |
GTGGCGCTCCTGTACCGTGGGTAAAGACCACCCACGG | 6558 |
|
195_207delinsTG-R |
CCGTGGGTGGTCTTTACCCACGGTACAGGAGCGCCAC | |
| 833T>C-F |
CTGCTTCCAGGTGCCGCCCCTGGACCAG | 6569 |
| 833T>C-R |
CTGGTCCAGGGGCGGCACCTGGAAGCAG | |
|
NR0B1-qPCR-F |
AGGGGACCGTGCTCTTTAAC | 214 |
|
NR0B1-qPCR-R |
ATGATGGGCCTGAAGAACAG | |
| β-actin-F |
GCCGGGACCTGACTGACTAC | 100 |
| β-actin-R |
TTCTCCTTAATGTCACGCACGAT | |
Plasmid construction and mutagenesis
The wild-type (WT) human NR0B1 and
NR5A1 complementary DNA (cDNA) clones were obtained from the
Dana-Farber/Harvard Cancer Center DNA Resource Core (Boston, MA,
USA). The NR0B1 cDNA was sub-cloned into pCSX/3Flag-DEST and
pLenti6.3/V5-DEST Gateway vectors (Invitrogen; Thermo Fisher
Scientific, Inc.) to generate the N-terminal Flag-tagged
NR0B1 and C-terminal V5-tagged NR0B1 expression
vectors, respectively. The NR5A1 cDNA was sub-cloned into
the pCSX/6HA-DEST Gateway vector. The NR0B1
c.195_207delinsTG and c.833T>C (Leu278Pro) mutations were
created using a PCR-based DpnI treatment method (21). The mutagenic primers employed in
the present study are listed in Table
I. Steroidogenic acute regulatory protein (StAR) promoter
sequences (-1,096 to +182) from human genomic DNA were cloned into
the pGL3-Enhancer vector (Promega Corp., Madison, WI, USA) to
generate a luciferase reporter plasmid (StAR-luc). All of the final
constructs were confirmed by DNA sequencing.
Cell culture and transient
transfection
The HEK293T human embryonic kidney cell line (which
does not express DAX1 protein) was obtained from the American Type
Culture Collection (Manassas, VA, USA) and maintained in Dulbecco's
modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.)
supplemented with 10% (v/v) fetal bovine serum (FBS; Thermo Fisher
Scientific, Inc.), 100 IU/ml penicillin and 100 µg/ml
streptomycin (Sigma-Aldrich, St. Louis, MO, USA) in a humidified
atmosphere with 5% CO2 atmosphere at 37°C. Transfection
was performed using Lipofectamine 3000 (Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions.
Purification of total RNA and
reverse-transcription quantitative PCR (RT-qPCR)
HEK293T cells were collected 24 h after
transfection. Total RNA was isolated using the RNeasy Mini Kit
(Qiagen) and then reverse-transcribed into cDNA. SYBR green-based
real-time qPCR was performed in a 96-well plate using FastStart
Universal SYBR Green Master (Roche, Basel, Switzerland) in a
StepOnePlus Real-Time PCR System (Invitrogen). The primers used for
qPCR of NR0B1 are listed in Table I and were purchased from Beijing
Genomics Institute (Shenzhen, China). The thermocycling protocol
was: 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and
60°C for 30 sec. Values were normalized to β-actin using the
2−ΔΔCq method (22).
Western blot analysis
HEK293T cells were seeded (5×105
cells/well) into six-well plates and transfected with N-terminal
Flag-tagged and C-terminal V5-tagged NR0B1 expression
vectors, respectively. Cytoplasmic and nuclear proteins were
extracted using a nuclear and cytoplasmic extraction kit (Pierce
Biotechnology, Inc., Rockford, IL, USA) at 24 h after transfection
according to the manufacturer's instructions. The protein
concentration was determined by the a Bicinchoninic Acid Protein
Assay kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions and 25 µg of cytoplasmic
and nuclear proteins were loaded into each lane. Cytoplasmic and
nuclear proteins were separated using 12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis Bio-Rad Laboratories, Inc.
(Hercules, CA, USA), and transferred onto polyvinylidene difluoride
membranes (Bio-Rad Laboratories, Inc.). The membranes were
incubated with blocking solution for 1 h at room temperature under
agitation. The blocking solution was Tris-buffered saline with
Tween-20 (TBST) buffer containing 5% non-fat milk. To make the 1X
TBST buffer, 2.423 g Tris Hcl and 8.006 g NaCl were dissolved in
800 ml ultra pure water, and 1 ml Tween-20 was added to adjust the
pH to 7.6. Subsequently, ultra pure water was added to make the
volume up to 1 litre. Tric Hcl, NaCl and Tween-20 were obtained
from Sigma-Aldrich. The membrane was probed with anti-Flag antibody
(Sigma-Aldrich; cat. no. F3165; 1:1,000) or anti-V5 antibody
(Abcam, Cambridge, MA, USA; cat. no. ab27671; 1:2,000),
anti-β-tubulin (cat. no. T5201, Sigma-Aldrich; 1:2,000) and anti
lamin B1 (Abcam; cat. no. ab133741; 1:5,000) for 1 h at room
temperature. β-tubulin and lamin B1 were used as cytoplasmic and
nuclear loading controls, respectively. The membranes were then
incubated with horseradish peroxidase (HRP)-conjugated
goat-anti-mouse IgG (cat. no. ab97023, Abcam; 1:5,000) and
HRP-conjugated goat-anti-rabbit IgG (cat. no. ab97051, Abcam;
1:5,000) for 1 h at room temperature. The membranes were washed 4
times in TBST buffer at room temperature under agitation, 5 min per
wash, to remove residual primary and secondary antibodies. The
blots were visualized using an enhanced chemiluminescnece Western
Blotting Substrate (Thermo Fisher Scientific, Inc.). The imaging
platform used was ImageQuant LAS 4000 mini (GE Healthcare,
Pittsburgh, PA, USA).
Luciferase assay
The regulatory role of DAX1 in modulating
transcription was monitored using the StAR-luc plasmid. In brief,
HEK293T cells were seeded into 96-well plates at a density of
6×104 cells/well and incubated for 24 h. The cells were
co-transfected with 20 ng StAR-luc, 0.5 ng pRL-SV40, and 60 ng WT
or mutant DAX1, and treated with 20 ng NR5A1 plasmid which encoded
steroidogenic factor-1 (SF-1). Total DNA was kept constant by
complementing each transfection with empty backbone plasmids
whenever it was necessary. Luciferase activities were measured from
cell lysates at 24 h post-transfection using the Dual-Glo
luciferase assay system (Promega Corp.) and normalized to Renilla
luciferase encoded by pRL-SV40. A reported NR0B1 mutation,
Leu278Pro, was used as the control (23).
Statistical analysis
Comparisons were made using the two-tailed Student's
t-test. Statistical analysis was conducted using Microsoft Excel
2013 (Microsoft Corporation, Redmond, WA, USA). Values are
expressed as the mean ± standard deviation from three independent
transient transfection assays. P<0.05 was considered to indicate
a statistically significant difference.
Results
Mutational analysis
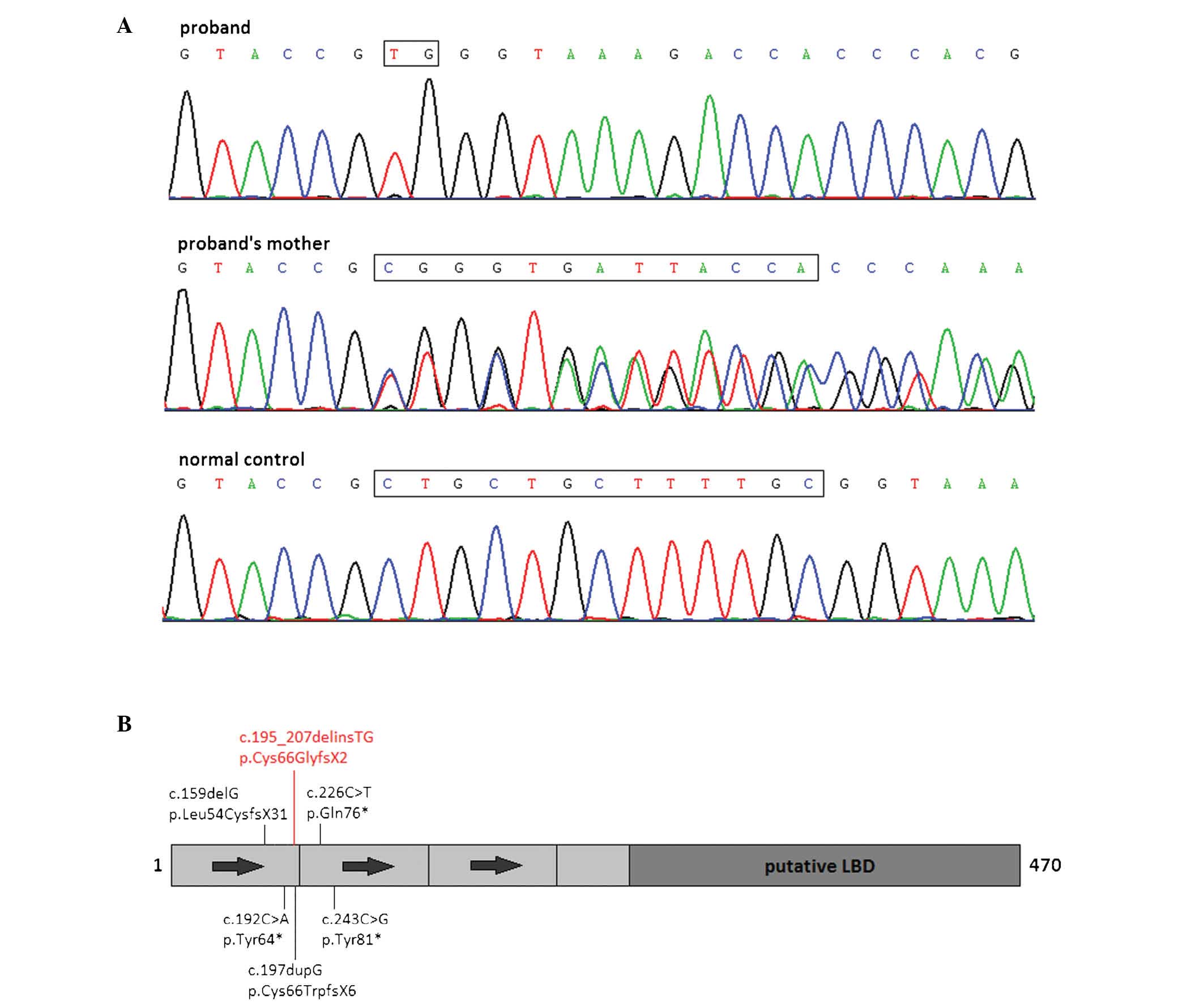
Direct sequencing of the proband revealed a small
indel variant in the NR0B1 gene, which was maternally
inherited (Fig. 2A). The indel
variant, c.195_207delinsTG, was located in exon 1 of the
NR0B1 gene and was predicted to cause a frameshift at amino
acid codon 66 (p.Cys66GlyfsX2, Fig.
2B) as predicted by the mutation analysis software named Alamut
Visual v.2.7.1 (Interactive Biosoftware, Rouen, France). This
variant has not been previously reported in the Human Genome
Mutation Database (http://www.hgmd.cf.ac.uk) or control databases such as
the 1,000 Genomes project (http://www.1000genomes.org), the National Heart, Lung
and Blood Institute Gene Ontology Exome Sequencing Project
(http://evs.gs.washington.edu/EVS) or the
Exome Aggregation Consortium (http://exac.broadinstitute.org), and therefore
represents a novel mutation, to the best of our knowledge.
Furthermore, the mutation was not detected in the 100
ethnicity-matched healthy control subjects.
The c.195_207delinsTG mutation in the
NR0B1 gene reduces DAX1 mRNA expression in vitro
To investigate the effect of the c.195_207delinsTG
mutation on NR0B1 transcription, WT and c.195_207delinsTG
mutant NR0B1 expression vectors were transiently and
individually transfected into HEK293T cells, followed by RNA
extraction and RT-qPCR analysis. DAX1 mRNA expression in the
NR0B1 mutant was significantly reduced (62% decrease;
P<0.001) compared to that in the NR0B1 WT group (Fig. 3).
The c.195_207delinsTG mutation in the
NR0B1 gene leads to low-level expression of truncated DAX1
protein
To investigate the effects of the c.195_207delinsTG
mutation on the synthesis and sub-cellular localization of DAX1,
N-terminal Flag-tagged and C-terminal V5-tagged NR0B1
expression vectors were transiently and individually transfected
into HEK293T cells, followed by extraction of cytoplasmic and
nuclear proteins, which were subjected to western blot analysis.
The WT DAX1 protein was ~55 kDa in length and localized to the
nucleus as well as the cytoplasm (Fig.
4). Compared to the levels of the WT DAX1 protein, the levels
of mutant DAX1 proteins in the transfected cells were markedly
lower. Application of the anti-Flag antibody showed that the
c.195_207delinsTG mutation encoded a truncated protein (~15 kDa),
which was predominantly located to the cytoplasm (Fig. 4A). Blotting with the anti-V5
antibody showed that the c.195_207delinsTG mutation encoded two
additional truncated proteins (~50 kDa) that were contained in the
nucleus as well as the cytoplasm (Fig.
4B).
The c.195_207delinsTG mutant of DAX1
shows enhanced suppression of SF-1-mediated StAR promoter
activity
DAX1 has been previously shown to inhibit
SF-1-mediated transactivation (16,17).
Therefore, a luciferase assay was performed in the present study to
investigate whether the c.195_207delinsTG mutation impaired the
repressor function of DAX1. As expected, WT DAX1 significantly
suppressed SF-1-mediated StAR promoter activity by 35.5±1.9%,
whereas the 'classic' AHC mutant, Leu278Pro, showed a complete lack
of repressive function. Compared to WT DAX1, the c.195_207delinsTG
mutant of DAX1 showed an even higher repressive function,
suppressing SF-1-induced StAR expression by 49.9±2.6% (Fig. 5).
Discussion
X-linked AHC with HHG is a rare genetic disorder
caused by mutations in the NR0B1 gene. A Chinese pedigree
was investigated in the present study. The proband, a 25-year-old
male, presented with adrenal insufficiency during childhood and
lack of development of secondary sexual characteristics during
puberty. Molecular analysis revealed a small indel mutation in the
NR0B1 gene. The diagnosis of X-linked AHC with HHG was
established based on clinical presentation, laboratory tests and
molecular analysis.
To date, >200 types of mutations of the
NR0B1 gene have been recorded in the Human Gene Mutation
Database. The majority of the mutations reported are small
deletions, followed by nonsense mutations and missense mutations.
More than half of the mutations are clustered at the carboxyl
terminus. To date, five small indel mutations in the NR0B1
gene, including c.273_274delinsT, c.585delinsCC, c.986_987delinsA,
c.1130delinsGT, and c.1376_1377delinsG have been described
(6,11,24–26).
These small indel mutations lead to a frameshift and cause
pre-mature truncation of the DAX1 protein. The present study
reported on a novel small indel mutation in the NR0B1 gene,
c.195_207delinsTG, which affects the amino terminus of the DAX1
protein. Several adjacent mutations, including c.159delG,
c.192C>A, c.197dupG, c.226C>T and c.243C>G, have been
reported to cause X-linked AHC (27–31).
The NR0B1 c.195_207delinsTG mutation is
predicted to cause frameshift and pre-mature termination. To
determine the effects of the c.195_207delinsTG mutation on DAX1
protein synthesis, transient expression studies were performed in
HEK293T cells transfected with vectors carrying WT or mutant DAX1
cDNA. RT-qPCR analysis showed that the levels of the transcript of
the NR0B1 c.195_207delinsTG mutant were significantly
reduced compared to those of the WT NR0B1 gene, indicating
that the aberrant NR0B1 mRNA with a pre-mature termination
codon (PTC) may trigger nonsense-mediated mRNA decay (NMD). NMD is
a well-known translation-coupled quality control system that
recognizes and leads to a reduction in PTC-harboring mRNAs
(32).
The decrease in DAX1 expression was also verified
using a western blot assay. Similar to the findings of other
studies (20,33), the WT DAX1 protein was about 55 kDa
in size and localized in the nucleus as well as the cytoplasm.
However, the NR0B1 c.195_207delinsTG mutation resulted in
various truncated DAX1 proteins. Western blot analysis using
antibodies specific for the N-terminal Flag-tag revealed the
presence of a truncated protein of ~15 kDa in length, which is the
p.Cys66GlyfsX2 mutant of DAX1 generated by the frameshift mutation
c.195_207delinsTG in the NR0B1 gene. The amount of
p.Cys66GlyfsX2 mutant of DAX1 was markedly lower than that of WT
DAX1. Furthermore, the p.Cys66GlyfsX2 mutant of DAX1 predominantly
located in the cytoplasm. It has been shown that LXXLL motifs and
an intact structure of the LBD are crucial for the nuclear
localization of DAX1 (34,35). However, the truncated
p.Cys66GlyfsX2 mutant of DAX1 only retained the first LXXLL motif,
which may have resulted in the abnormal intracellular distribution
of the mutant DAX1. Furthermore, this severely truncated DAX1
protein was probably non-functional. Using the C-terminal V5-tag,
two additional truncated ~50-kDa proteins that were present in the
nucleus as well as in the cytoplasm were detected. Based on a
previous in vitro study, an amino-truncated isoform of DAX1,
which is normally expressed as a minor protein variant with a
partially preserved repressor function, can be generated from an
alternate in-frame translation start site (methionine, codon 83)
(36). As the alternate
translation initiation site is located downstream of the
NR0B1 c.195_207delinsTG mutation, the formation of this
isoform was not affected by the indel mutation. This infers that
the lower band, which was also present at low levels in WT DAX1, is
the amino-truncated isoform of DAX1. In addition, a
higher-molecular weight band was observed in the mutant DAX1 only.
Further study is required to determine whether this
higher-molecular weight protein resulted from aberrantly spliced
mutant mRNA or protein with additional modification induced by the
NR0B1 c.195_207delinsTG mutation.
DAX1 has been shown to be a negative regulator,
which acts by repressing the SF-1-mediated transactivation of
various genes involved in steroidogenesis, such as StAR (16,17).
Loss of this inhibitory property in DAX1 through NR0B1
mutations was demonstrated to be responsible for the pathology of
X-linked AHC and HHG (3,16,20,23).
To assess the effects of the c.195_207delinsTG mutation of
NR0B1 on the repressor function of DAX1, a luciferase assay
was performed. The results unexpectedly showed that this mutation
did not impair the repressor function of DAX1. Instead, compared to
the WT DAX1, the c.195_207delinsTG mutant of DAX1 displayed an even
greater ability to suppress SF-1-induced StAR expression. Although
the DAX1 Gln37Term and Trp39Term mutations were reported to have a
milder phenotype due to the expression of a partially functional,
amino-truncated DAX-1 protein, other naturally occurring nonsense
or frameshift NR0B1 mutations that cause PTC upstream of the
putative alternative start site are associated with the classical
AHC phenotype (36). These
findings suggested that the mutations of NR0B1 leading to
X-linked AHC with HHG are not uniformly associated with the loss of
function or DAX1 and are likely to involve molecular functions
other than its role as a repressor of SF-1 activation.
In conclusion, the present study reported on a
Chinese pedigree including a case of X-linked AHC and HHG. A novel
small indel mutation, c.195_207delinsTG, in the NR0B1 gene
was identified in the proband. In vitro studies showed that
the mutation reduced DAX1 expression and resulted in N-terminal
truncated as well as C-terminal truncated DAX1 protein.
Furthermore, a Luciferase assay demonstrated that the mutation
enhanced the repressor activity of DAX1. The present study
indicated the complexity of the underlying mechanisms of the
association of NR0B1 with X-linked AHC and HHG. Further
in vivo studies will help elucidate the biological effects
of mutant DAX1 in the development and function of the adrenal gland
and hypothalamic-pituitary-gonadal axis.
Acknowledgments
The authors would like to thank all participants of
the present study.
References
|
1
|
MacMahon HE, Wagner R and Weiner DB: Acute
adrenal insufficiency due to congenital defect. Am J Dis Child.
94:282–285. 1957.
|
|
2
|
Zanaria E, Muscatelli F, Bardoni B, Strom
TM, Guioli S, Guo W, Lalli E, Moser C, Walker AP, McCabe ER, et al:
An unusual member of the nuclear hormone receptor superfamily
responsible for X-linked adrenal hypoplasia congenita. Nature.
372:635–641. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reutens AT, Achermann JC, Ito M, Ito M, Gu
WX, Habiby RL, Donohoue PA, Pang S, Hindmarsh PC and Jameson JL:
Clinical and functional effects of mutations in the DAX-1 gene in
patients with adrenal hypoplasia congenita. J Clin Endocrinol
Metab. 84:504–511. 1999.PubMed/NCBI
|
|
4
|
Lin L, Gu WX, Ozisik G, To WS, Owen CJ,
Jameson JL and Achermann JC: Analysis of DAX1 (NR0B1) and
steroidogenic factor-1 (NR5A1) in children and adults with primary
adrenal failure: Ten years' experience. J Clin Endocrinol Metab.
91:3048–3054. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uttley WS: Familial congenital adrenal
hypoplasia. Arch Dis Child. 43:724–730. 1968. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Habiby RL, Boepple P, Nachtigall L, Sluss
PM, Crowley WF Jr and Jameson JL: Adrenal hypoplasia congenita with
hypogonadotropic hypogonadism: Evidence that DAX-1 mutations lead
to combined hypothalamic and pituitary defects in gonadotropin
production. J Clin Invest. 98:1055–1062. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zachmann M, Illig R and Prader A:
Gonadotropin deficiency and cryptorchidism in three prepubertal
brothers with congenital adrenal hypoplasia. J Pediatr. 97:255–257.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hammond J, Howard NJ, Brookwell R,
Purvis-Smith S, Wilcken B and Hoogenraad N: Proposed assignment of
loci for X-linked adrenal hypoplasia and glycerol kinase genes.
Lancet. 1:541985. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bartley JA, Patil S, Davenport S,
Goldstein D and Pickens J: Duchenne muscular dystrophy, glycerol
kinase deficiency and adrenal insufficiency associated with Xp21
interstitial deletion. J Pediatr. 108:189–192. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Francke U, Harper JF, Darras BT, Cowan JM,
McCabe ER, Kohlschütter A, Seltzer WK, Saito F, Goto J, Harpey JP,
et al: Congenital adrenal hypoplasia, myopathy and glycerol kinase
deficiency: Molecular genetic evidence for deletions. Am J Hum
Genet. 40:212–227. 1987.PubMed/NCBI
|
|
11
|
Muscatelli F, Strom TM, Walker AP, Zanaria
E, Récan D, Meindl A, Bardoni B, Guioli S, Zehetner G, Rabl W, et
al: Mutations in the DAX-1 gene give rise to both X-linked adrenal
hypoplasia congenita and hypogonadotropic hypogonadism. Nature.
372:672–676. 1994. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo W, Burris TP, Zhang YH, Huang BL,
Mason J, Copeland KC, Kupfer SR, Pagon RA and McCabe ER: Genomic
sequence of the DAX1 gene: An orphan nuclear receptor responsible
for X-linked adrenal hypoplasia congenital and hypogonadotropic
hypogonadism. J Clin Endocrinol Metab. 81:2481–2486.
1996.PubMed/NCBI
|
|
13
|
Guo W, Burris TP and McCabe ER: Expression
of DAX1, the gene responsible for X-linked adrenal hypoplasia
congenita and hypogonadotropic hypogonadism, in the
hypo-thalamic-pituitary-adrenal/gonadal axis. Biochem Mol Med.
56:8–13. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mukai T, Kusaka M, Kawabe K, Goto K,
Nawata H, Fujieda K and Morohashi K: Sexually dimorphic expression
of Dax-1 in the adrenal cortex. Genes Cells. 7:717–729. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikeda Y, Swain A, Weber TJ, Hentges KE,
Zanaria E, Lalli E, Tamai KT, Sassone-Corsi P, Lovell-Badge R,
Camerino G and Parker KL: Steroidogenic factor 1 and Dax-1
colocalize in multiple cell lineages: Potential links in endocrine
development. Mol Endocrinol. 10:1261–1272. 1996.PubMed/NCBI
|
|
16
|
Ito M, Yu R and Jameson JL: DAX-1 inhibits
SF-1-mediated transactivation via a carboxy-terminal domain that is
deleted in adrenal hypoplasia congenital. Mol Cell Biol.
17:1476–1483. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zazopoulos E, Lalli E, Stocco DM and
Sassone-Corsi P: DNA binding and transcriptional repression by
DAX-1 blocks steroidogenesis. Nature. 390:311–315. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suzuki T, Kasahara M, Yoshioka H,
Morohashi K and Umesono K: LXXLL-related motifs in Dax-1 have
target specificity for the orphan nuclear receptors Ad4BP/SF-1 and
LRH-1. Mol Cell Biol. 23:238–249. 2003. View Article : Google Scholar :
|
|
19
|
Desmangles JC, Lappe JM, Lipaczewski G and
Haynatzki G: Accuracy of pubertal Tanner staging self-reporting. J
Pediatr Endocrinol Metab. 19:213–221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li S and Wilkinson MF: Site-directed
mutagenesis: A two-step method using PCR and DpnI. Biotechniques.
23:588–590. 1997.PubMed/NCBI
|
|
21
|
Li N, Liu R, Zhang H, Yang J, Sun S, Zhang
M, Liu Y, Lu Y, Wang W, Mu Y, et al: Seven novel DAX1 mutations
with loss of function identified in Chinese patients with
congenital adrenal hypoplasia. J Clin Endocrinol Metab.
95:E104–E111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Achermann JC, Ito M, Silverman BL, Habiby
RL, Pang S, Rosler A and Jameson JL: Missense mutations cluster
within the carboxyl-terminal region of DAX-1 and impair
transcriptional repression. J Clin Endocrinol Metab. 86:3171–5.
2001.PubMed/NCBI
|
|
24
|
Peter M, Viemann M, Partsch CJ and Sippell
WG: Congenital adrenal hypoplasia: Clinical spectrum, experience
with hormonal diagnosis and report on new point mutations of the
DAX-1 gene. J Clin Endocrinol Metab. 83:2666–2674. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Esden-Tempska Z, Lewczuk A, Tobias ES,
Borozdin W, Kohlhase J and Sworczak K: Delayed diagnosis of adrenal
hypoplasia congenita in a patient with a new mutation in the NR0B1
gene. J Pediatr Endocrinol Metab. 25:147–148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nakae J, Tajima T, Kusuda S, Kohda N,
Okabe T, Shinohara N, Kato M, Murashita M, Mukai T, Imanaka K and
Fujieda K: Truncation at the C-terminus of the DAX-1 protein
impairs its biological actions in patients with X-linked adrenal
hypoplasia congenita. J Clin Endocrinol Metab. 81:3680–3685.
1996.PubMed/NCBI
|
|
27
|
Tsai WY and Tung YC: Novel deletion
mutations of the DAX1 (NR0B1) gene in two Taiwanese families with
X-linked adrenal hypoplasia congenita. J Pediatr Endocrinol Metab.
18:991–997. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Domenice S, Latronico AC, Brito VN,
Arnhold IJ, Kok F and Mendonca BB: Adrenocorticotropin-dependent
precocious puberty of testicular origin in a boy with X-linked
adrenal hypoplasia congenita due to a novel mutation in the DAX1
gene. J Clin Endocrinol Metab. 86:4068–4071. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wheeler B, George PM, Mackenzie K, Hunt P,
Potter HC and Florkowski CM: Three cases of congenital adrenal
hypoplasia with novel mutations in the (NROB1) DAX-1 gene. Ann Clin
Biochem. 45:606–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
García-Malpartida K, Gómez-Balaguer M,
Solá-Izquierdo E, Fuentes-Pardilla MJ, Jover-Fernández A, Sanz-Ruiz
I and Hernández-Mijares A: A novel mutation in DAX1 (NR0B1) causing
X-linked adrenal hypoplasia congenita: Clinical, hormonal and
genetic analysis. Endocrine. 36:275–280. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang YH, Huang BL, Anyane-Yeboa K,
Carvalho JA, Clemons RD, Cole T, De Figueiredo BC, Lubinsky M,
Metzger DL, Quadrelli R, et al: Nine novel mutations in NR0B1
(DAX1) causing adrenal hypoplasia congenita. Hum Mutat. 18:5472001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brogna S and Wen J: Nonsense-mediated mRNA
decay (NMD) mechanisms. Nat Struct Mol Biol. 16:107–113. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lalli E, Ohe K, Hindelang C and
Sassone-Corsi P: Orphan receptor DAX-1 is a shuttling RNA binding
protein associated with polyribosomes via mRNA. Mol Cell Biol.
20:4910–4921. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kawajiri K, Ikuta T, Suzuki T, Kusaka M,
Muramatsu M, Fujieda K, Tachibana M and Morohashi K: Role of the
LXXLL-motif and activation function 2 domain in subcellular
localization of Dax-1 (dosage-sensitive sex reversal-adrenal
hypoplasia congenita critical region on the X chromosome, gene 1).
Mol Endocrinol. 17:994–1004. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lehmann SG, Lalli E and Sassone-Corsi P:
X-linked adrenal hypoplasia congenita is caused by abnormal nuclear
localization of the DAX-1 protein. Proc Natl Acad Sci USA.
99:8225–8230. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ozisik G, Mantovani G, Achermann JC,
Persani L, Spada A, Weiss J, Beck-Peccoz P and Jameson JL: An
alternate translation initiation site circumvents an amino-terminal
DAX1 nonsense mutation leading to a mild form of X-linked adrenal
hypoplasia congenital. J Clin Endocrinol Metab. 88:417–423. 2003.
View Article : Google Scholar : PubMed/NCBI
|