Introduction
Fetal growth restriction is a common complication of
pregnancy, and a significant cause of perinatal morbidity and
mortality (1). Numerous studies
have reported that adverse conditions during critical periods of
development can alter physiological processes leading to metabolic
diseases, including type 2 diabetes, hypertension, fatty liver
disease and cardiovascular disease (2,3).
Furthermore, the majority of small for gestational age (SGA)
offspring exhibit compensatory growth during the first 2 years of
life, which may contribute to a higher body fat mass from as young
as 2–12 months of age (4), and
increased body fatness and abdominal fat accumulation during
childhood (5) and adulthood
(6,7). Considering the social and economic
burden of chronic metabolic disease, it is important to elucidate
the underlying mechanisms and provide potential strategies for the
prevention of long-term metabolic consequences in SGA
offspring.
Lipoprotein lipase (LPL), which is a key lipid
metabolism enzyme that hydrolyzes triglyceride (TG), provides free
fatty acids (FFAs) for cells and affects the maturation of
circulating lipoproteins (8,9). LPL
has its own developmental genetic program, the activity and
expression of which can vary greatly between tissues. In the liver
tissue of fetal and neonatal rats high LPL activity has been
detected (10); however, the
expression of LPL progressively decreases and falls to nearly
undetectable levels by the time of weaning (11), as determined by measuring
age-related decreases in LPL activity, LPL synthesis and LPL mRNA
expression (12). Numerous studies
have suggested that, in the pathological state, LPL expression may
undergo alterations resulting in various diseases, including
atherosclerosis, obesity and diabetes (13–15).
The present study aimed to determine whether hepatic LPL gene
expression was altered in SGA male rat offspring, and to
investigate the potential mechanisms underlying expression
alterations.
Several nuclear receptors can activate the
transcription of LPL, including peroxisome proliferator-activated
receptors, sterol regulatory element-binding protein-1c and liver X
receptors (LXRs) (13,16,17).
LXRs are nuclear receptors involved in the transcriptional
regulation of de novo TG synthesis. Two isoforms of LXR:
Liver X receptor-α (LXR-α) and LXR-β, have been identified in birds
and mammals. As a more selective regulator of LPL than LXR-β, LXR-α
binds to LXR response elements (LXRE), which contain a hexameric
nucleotide direct repeat spaced by four bases (DR4), in the LPL
promoter in order to govern regulation following activation by
oxysterols (17). A previous study
demonstrated that maternal protein restriction can alter rat LXR-α
expression and lead to long-term epigenetic alterations in LXR
target genes associated with lipid homeostasis (18). The present study hypothesized that
maternal undernutrition during pregnancy-induced SGA male offspring
would exhibit alterations in the expression of LPL, which may be
mediated by increased or decreased binding of LXR-α to the LPL gene
promoter.
Epigenetics serves a critical function in affecting
gene transcription. Previous studies have focused on the
identification of epigenetic dysregulation at the promoters of
certain genes in SGA offspring. Park et al (19) demonstrated that histone
modifications are involved in the effects of uteroplacental
insufficiency on islet pancreatic and duodenal homeobox gene 1
transcription in rats. Sohi et al (18) indicated that maternal protein
restriction leads to long-term decreases in histone H3 lysine (K)9
(H3K9) and H3K14 surrounding the promoter of the LXR target gene
cholesterol 7α-hydroxylase, resulting in hypercholesterolemia in
SGA offspring. The present study aimed to determine whether
post-translational histone modifications may also influence the
expression of LPL in SGA male rat offspring.
Materials and methods
Animal model
Animal experiments were performed at the Laboratory
Animal Center of Zhejiang University (Hangzhou, China). All animal
experimental procedures were approved by the Animal Ethics
Committee of Zhejiang University, School of Medicine.
Briefly, 24 Sprague Dawley rats (16 male and 8
female; SLRC Laboratory Animal Co., Ltd., Shanghai, China) were
housed under standard conditions (room temperature, 20–22°C;
humidity, 40–60%). After 1 week of acclimation, male and female
rats were mated overnight, and the presence of sperm in a vaginal
smear was designated as gestational day 1. Pregnant rats were
arbitrarily divided into two groups: The control group continued to
receive an ad libitum chow diet; the second group was
subjected to food restriction and received 50% of their usual daily
intake until parturition. The pregnant rats delivered
spontaneously, and the litter size was randomly culled to eight per
mother at birth, in order to assure uniformity of litter size
between the SGA and appropriate for gestational age (AGA) litters.
All female offspring, as well as male offsprings that did not
meeting the criteria for AGA and SGA, were culled. The criteria for
AGA were as follows: Offspring of normal intake and birth weight
between mean ± standard deviation (SD). The criteria for SGA were
as follows: Offspring of food-restricted mothers and birth weight
<-2 SD of the AGA group. Following parturition, mothers from the
food restricted group were sacrificed by the administration of 20
ml chloral hydrate (J&I Biological, Shanghai, China). The pups
were cross-fostered from food-restricted mothers to ad
libitum-fed mothers. Both groups were given ad libitum
access to food. At 1 day and 3 weeks of age, the rats were
sacrificed by anesthesia (chloral hydrate; dose, 2 ml 1-day-old
rats and 10 ml for 3-week-old rats). The ad libitum mothers
were sacrificed by the administration of 20 ml chloral hydrate.
Liver tissue samples were harvested and snap-frozen in liquid
nitrogen for subsequent processing.
Hepatic TG content
Total hepatic TG content was determined using a
GPO-PAP enzymatic assay (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Briefly, liver tissue (30–50 mg) was
homogenized in ethanol (9 ml ethanol, 1 g liver sample), the
mixture was microfuged at 664 × g for 10 min at 4°C, and the
supernatant was transferred to new tubes. The reaction system was
prepared according to the manufacturer's instructions. The GPO-PAP
enzymatic assay protocol includes the following steps: First, the
lipids are broken down via the hydrolysis of triglycerides into
glycerol and FFAs. Then the glycerol is converted to glycerol
3-phosphate via adenosine triphosphate and glycerol kinase, and is
further converted to dihydroxyacetone phosphate and hydrogen
peroxide via glycerophosphate oxidase. Under the effect of
peroxidase, red quinones are produced when hydrogen peroxide meets
4-amino-antipyrine and 4-chlorophenol. The color degree of quionoes
is propotional to triglyceride concentration. Following a 5 min
incubation at 37°C, the final mixtures in 96-cell plates were
rapidly quantified at 500 nm using a micro-plate reader (Varioskan
Flash 3001, Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Quantification of TG content was based on the following
calculation: {[Sample optical density (OD) value-blank OD
value]/(calibration OD value-blank OD value)} × calibration
concentration. Finally, data were expressed as mg/g of liver.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Briefly, total RNA was isolated from the liver
tissue samples using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). High-Capacity cDNA Reverse Transcription
kit (Applied Biosystems; Thermo Fisher Scientific, Inc.) was used
to reverse transcribe 2 µg total RNA using the following
program: 25°C for 10 min, 37°C for 120 min, 85°C for 5 min, and a
4°C hold. To measure the relative mRNA expression levels, RT-qPCR
was performed using an Applied Biosystems StepOnePlus™ Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The PCR cycling conditions were as follows: 94°C for 10 min,
followed by 40 cycles at 94°C for 20 sec and 60°C for 1 min.
RT-qPCR was performed using SYBR® Select Master Mix
(Applied Biosystems; Thermo Fisher Scientific, Inc.) All reactions
were performed in triplicate. The relative mRNA expression levels
were calculated using the 2−ΔΔCq method (20). The primer sequences were as
follows: LXR-α, forward 5′-GAGAGCATCACCTTCCTCAAG-3′, reverse
5′-TCATGGATCTGGAGAACTCAAAG-3′; LPL, forward
5′-ACAGGTGCAATTCCAAGGAG-3′, reverse 5′-CTTTCAGCCACTGTGCCATA-3′; and
glyceraldehyde 3-phosphate dehydrogenase (GAP DH), forward
5′-GACAACTTTGGCATCGTGGA-3′ and reverse: 5′-ATGCAGGGATGATGTTCTGG-3′.
Primers were purchased from Thermo Fisher Scientific, Inc.
Western blotting
The liver tissue samples were homogenized in lysis
buffer (Beyotime Institute of Biotechnology, Hangzhou, China) and
centrifuged at 12,000 × g for 10 min at 4°C. The protein
concentration was determined using a Bicinchoninic Acid Protein
Assay kit (Beyotime Institute of Biotechnology). Equal amounts of
protein (80 µg) were separated by 10% sodium dodecyl sulfate
(SDS)-polyacrylamide gel electrophoresis and were electroblotted
onto a polyvinylidene difluoride membrane (0.2 µm pore size;
EMD Millipore, Billerica, MA, USA). The membrane was blocked in 5%
non-fatmilk for 2 h at room temperature. Following blocking, the
membrane was incubated at 4°C with anti-LXR-α [dilution 1:3,000;
cat. no. ab41902; Abcam (Hong Kong) Ltd., Hong Kong, China),
anti-LPL (dilution 1:200; cat. no. sc-32885; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and anti-GAPDH antibodies
(dilution 1:5,000; cat. no. 5174; Cell Signaling Technology, Inc.,
Danvers, MA USA) for 12 h. The blots were analyzed by ImageJ
version 1.39 software (National Institutes of Health, Bethesda, MD,
USA). Subsequently, the membrane was incubated with horseradish
peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG)
(HRP-labeled (cat. no., A0208; dilution 1:2,000; Beyotime Institute
of Biotechnology) and HRP-labeled goat anti-mouse IgG secondary
antibodies (cat. no., A0216; dilution 1:2,000; Beyotime Institute
of Biotechnology) for 2 h at room temperature. Signals were
detected using enhanced chemiluminescence, according to the
manufacturer's protocol (SuperSignal chemiluminescent substrates;
Pierce; Thermo Fisher Scientific, Inc.).
Chromatin immunopreciptation (ChIP)
The ChIP assay was performed according to the
manufacturer's protocol (EZ-ChIP kit; EMD Millipore). Liver tissue
samples (40 mg) were fixed in 1% formaldehyde for 10 min at room
temperature. Cross-linking was terminated by the addition of
glycine (1 M). Following two washes with cold phosphate-buffered
saline, the liver tissue was resuspended in 1 ml SDS lysis buffer
supplemented with 5 µl 1X protease inhibitor cocktail II.
The lysates were aliquoted to 300–400 µl per microfuge tube
and were sonicated on ice, in order to shear the DNA to a length
between 200 and 1,000 bp. The sheared crosslinked chromatin was
diluted 10-fold in dilution buffer containing protease inhibitor
cocktail II. The chromatin solution was pre-cleared with 60
µl protein G agarose at 4°C for 1 h with rotation. The
chromatin solutions were then microfuged at 4,000 × g for 1 min at
4°C to pellet agarose, and the supernatant was placed in new tubes,
with 10 µl removed as input. The supernatant fractions were
incubated overnight on a rocking platform with antibodies against
acetylated histone H3K9 [4 µg; cat. no. ab10812; Abcam (Hong
Kong) Ltd.], acetylated histone H3K14 [4 µg, cat. no.
ab52946; Abcam (Hong Kong) Ltd.] and ChIP-grade LXR-α [5 µg;
cat. no. ab41902; Abcam (Hong Kong) Ltd.] at 4°C. Subsequently, 60
µl protein G agarose was added to the tubes, which were
incubated on a rocking platform for 1 h at 4°C. Following
centrifugation at 4,000 × g at 4°C for 1 min, the agarose beads
containing the immunoprecipitated complexes were washed
sequentially in Low Salt Immune Complex Wash Buffer, High Salt
Immune Complex Wash Buffer, LiCI Immune Complex Wash Buffer, and 2X
TE buffer. The immune complexes were eluted twice with 200
µl elution buffer (10 µl 20% SDS, 20 µl 1 M
NaHCO3, 170 µl sterile distilled water) at room
temperature. Elution buffer (200 µl) was also added to the
input tubes. Subsequently, 8 µl 5 M NaCl was added to the
elute and the cross-linking of the immunoprecipitated chromatin
complexes and input controls were reversed by heating at 65°C for 5
h. Following treatment with Proteinase K, Tris-HCl and EDTA for 2 h
at 45°C, the DNA was purified, according to the protocol of the
manufacturer of the EZ-CHIP kit (EMD Millipore, Billerica, MA,
USA).
The putative LXR-binding site in the promoter region
of LPL was determined using MatInspector Software (http://www.genomatix.de/online_help/help_matinspector/matinspector_help.html).
Primers were purchased from Invitrogen (Thermo Fisher Scientific,
Inc.) and were as follows: Forward (5′-ATTCTCCACCTTGTCCCTTTG-3′)
and reverse (5′-GCTTGATTCCCAGAACCCAC-3′) primers that amplify −3438
to −3423 promoter regions encompassing the rat LPL LXRE site
(GAGGCC_DR4_GAGGGC), and primers (promoter A1, forward
5′-TCTGCTTTGCTGCTGGAACT-3′, reverse 5′-AGACGAAACGACGACCTTGA-3′;
promoter A2, forward 5′-CACTGTAACGAGGCTCAACG-3′, reverse
5′-GTGACATTGCTCCGAGTTGC-3′; and promoter A3, forward
5′-GAGGCAGAAAGTCATGGTCAAATA-3′ and reverse
5′-CTCCGTCTTTCAGTACCAGTTTAT-3′) surrounding the promoter were used
to examine the acetylation status of acetylation of histone H3 (K9,
K14) at the promoter of LPL. For negative controls, ChIP assays
were performed using an immunoglobulin G antibody (1 µg;
part of the EZ-ChIP kit) to determine the immuno-specificity of the
antibodies for the LPL promoter. The DNA samples from the input,
unbound and bound fractions were determined by qPCR, according to
the previously described protocol. The relative abundance of the
immunoprecipitated chromatin, as compared with the input chromatin
was determined using the 2−ΔΔCq method (20).
Statistical analysis
SPSS 18.0 (SPSS, Inc., Chicago, IL, USA) was used
for data analysis. All data are expressed as the mean ± standard
error of the mean. Statistical significance was calculated using
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
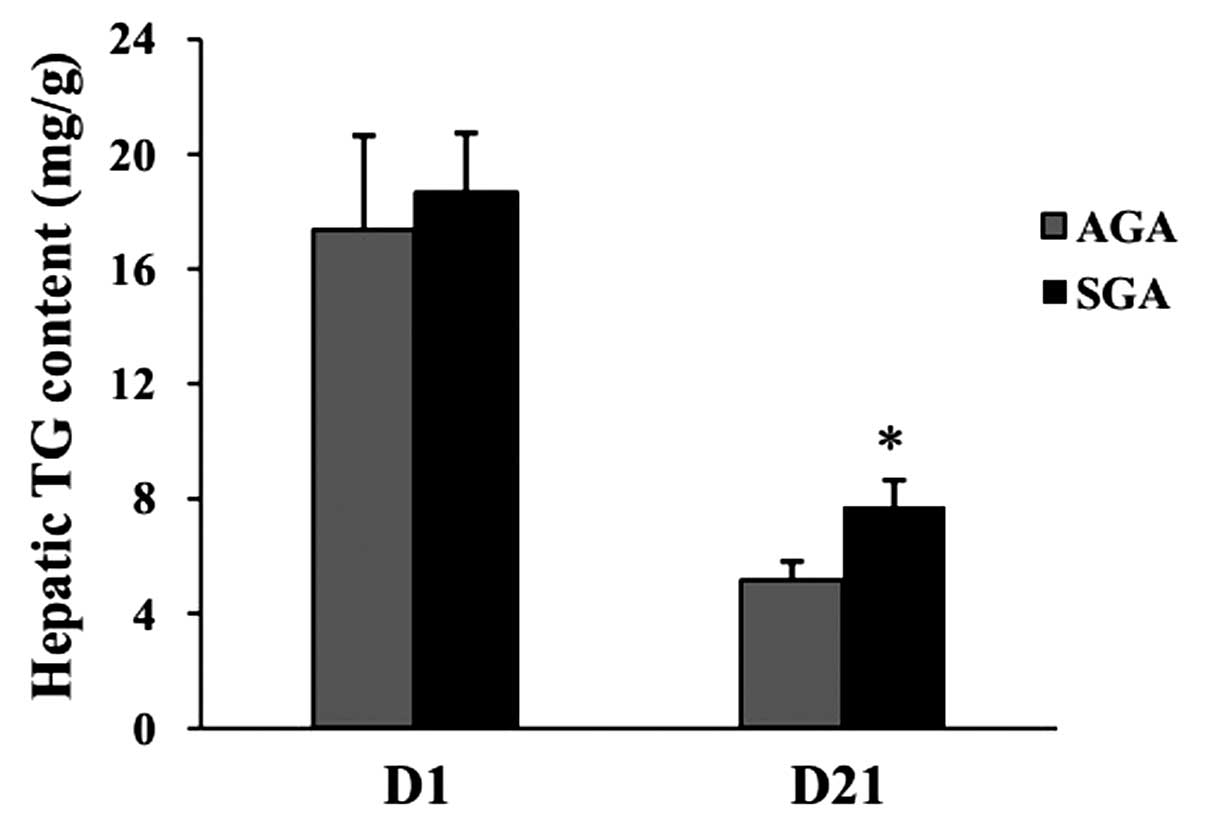
Hepatic TG content
At 1 day of age, no differences were evident in
hepatic TG levels between the AGA and SGA male rats (AGA,
17.39±3.25 mg/g; SGA, 18.68±2.05 mg/g; P>0.05) (Fig. 1). At 3 weeks of age, the hepatic TG
levels were increased in the SGA male rats (AGA, 5.18±0.63 mg/g;
SGA, 7.73±0.91 mg/g; P<0.05), as compared with the AGA rats.
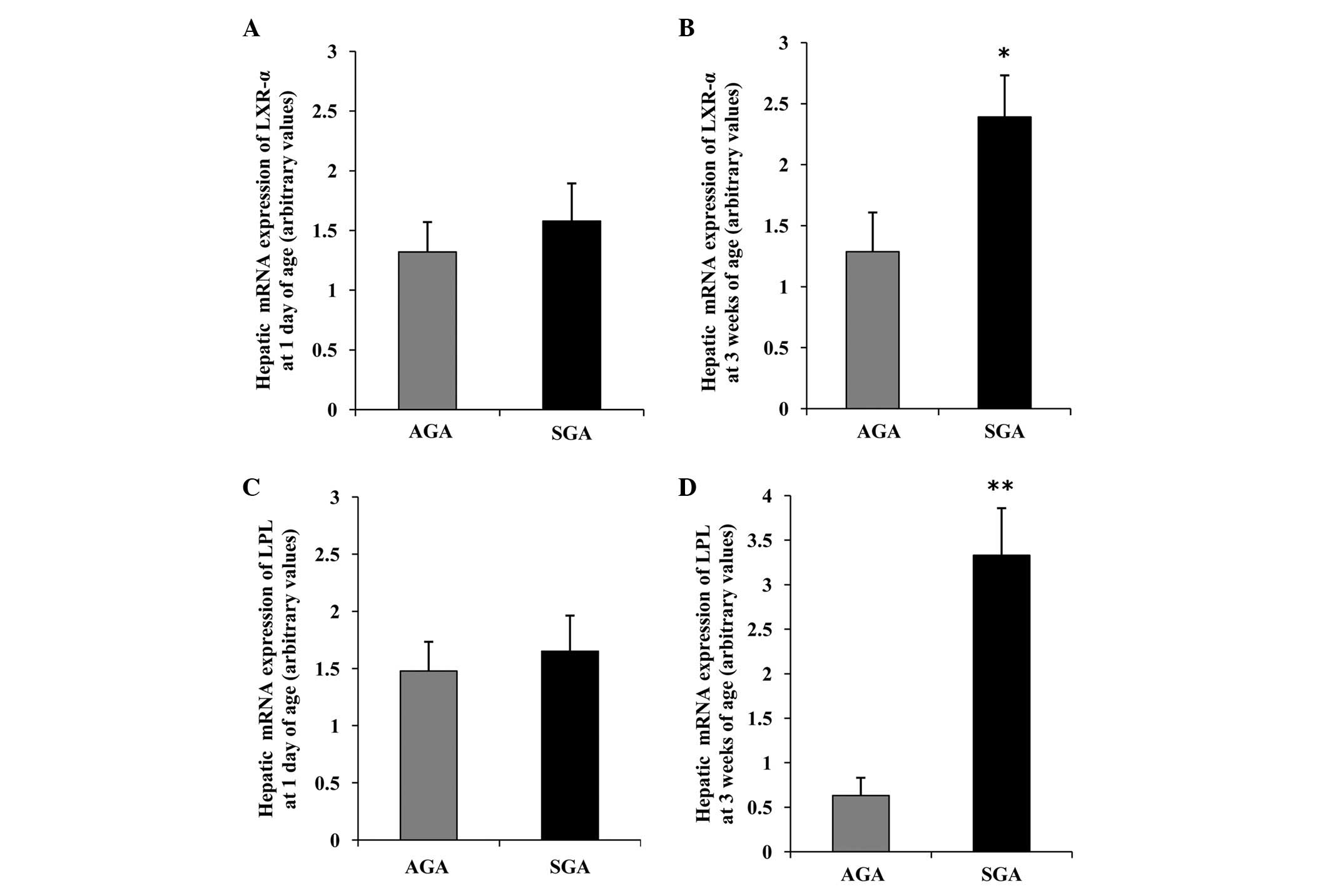
Hepatic LXR-α mRNA expression levels are
increased, concomitant with an increase in LPL mRNA in 3-week-old
SGA rats
The hepatic mRNA expression patterns in both groups
are presented in Fig. 2. At 1 day
of age, there was no difference in the mRNA expression levels of
LXR-α and LPL between the AGA and SGA male rats (Fig. 2A and C). However, at 3 weeks of
age, the SGA male rats had significantly increased mRNA expression
levels of LXR-α and LPL, as compared with the AGA rats (P<0.05
and P<0.01, respectively) (Fig. 2B
and D).
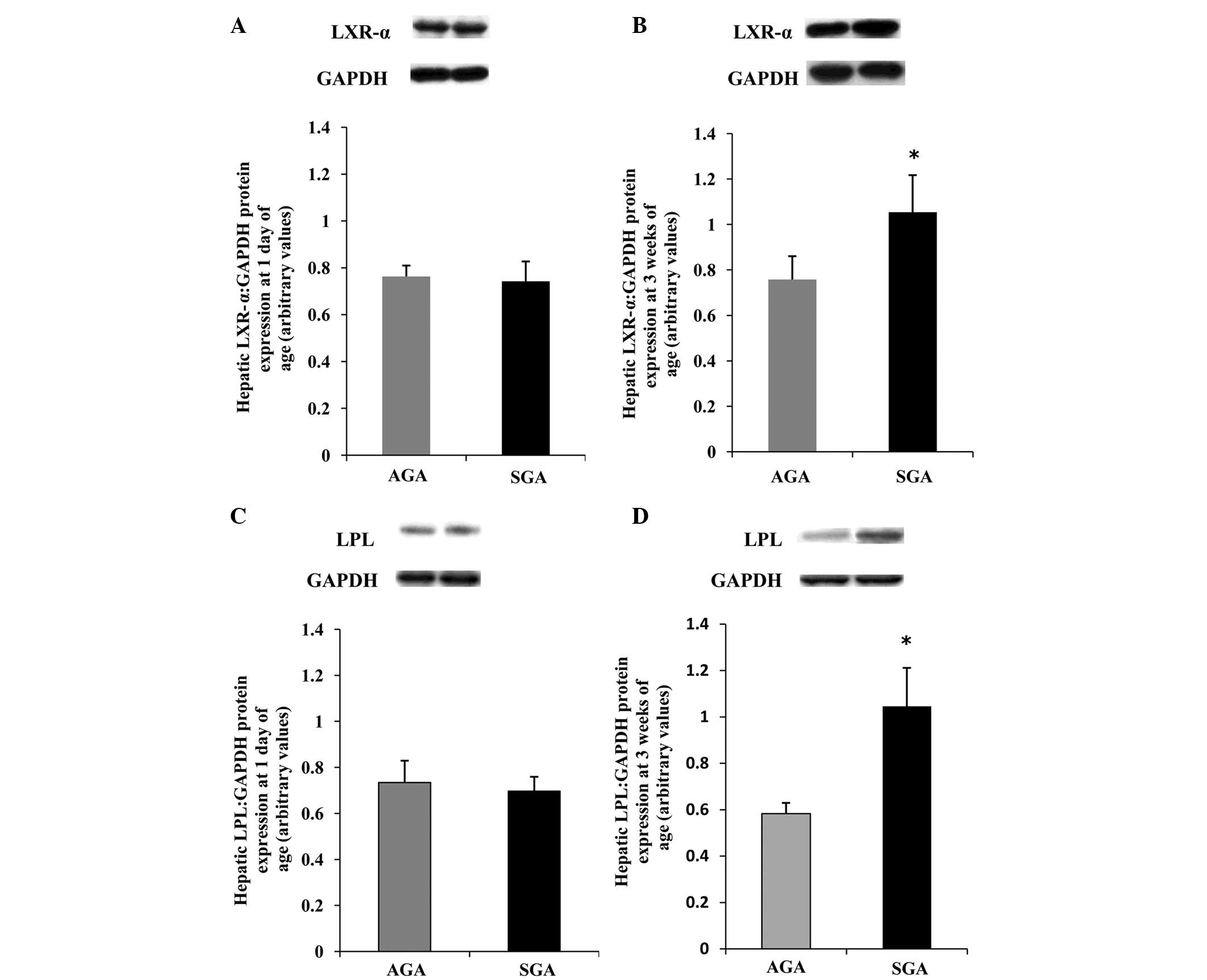
Hepatic LXR-α protein expression levels
are increased, concomitant with an increase in LPL protein in
3-week-old SGA rats
To obtain further information regarding the
differences in protein expression, western blotting was performed
(Fig. 3). The protein expression
levels of LXR-α and LPL were increased (P<0.05) in the SGA male
rats, as compared with the AGA rats at 3 weeks of age (Fig. 3B and D); however, no differences
were detected between the SGA and AGA male rats at 1 day of age
(Fig. 3A and C).
LXR-α binding to the LXRE in the promoter
regions of LPL is increased in 3-week-old SGA rats
To determine whether there were alterations in the
recruitment of LXR-α to the promoter regions of LPL containing a
well-characterized LXRE site, ChIP analyses were performed with
antibodies specific for LXR-α. Negative controls demonstrated that
the immunoprecipitations were specific for the indicated
antibodies. At 3 weeks of age, the SGA male rats exhibited a marked
increase in the binding of LXR-α to the promoter regions of LPL, as
compared with in the AGA rats (P<0.05) (Fig. 4).
Acetylation of lysine residues 9 and 14
on histone H3 surrounding the promoter regions of LPL is increased
in 3-week-old SGA rats
ChIP was further used to investigate whether
chromatin remodeling could be a factor influencing the observed
increase in LPL mRNA and protein expression levels in 3-week-old
male SGA rats. Three sites (A1: −87 to +177; A2: −136 to −533 and
A3: −644 to −896) along the LPL promoter were analyzed. The hepatic
levels of acetylated histone H3K9 in the LPL promoter A1, A2 and A3
regions of SGA rats were increased 1.71-fold, 2.73-fold and
2.50-fold, respectively (P<0.05), as compared with the AGA rats
(Fig. 5A). The hepatic levels of
acetylated histone H3K14 in the LPL promoter A1, A2 and A3 regions
of SGA rats were increased 1.69-fold, 1.86-fold and 2.67-fold,
respectively (P<0.05), as compared with the AGA rats (Fig 5B).
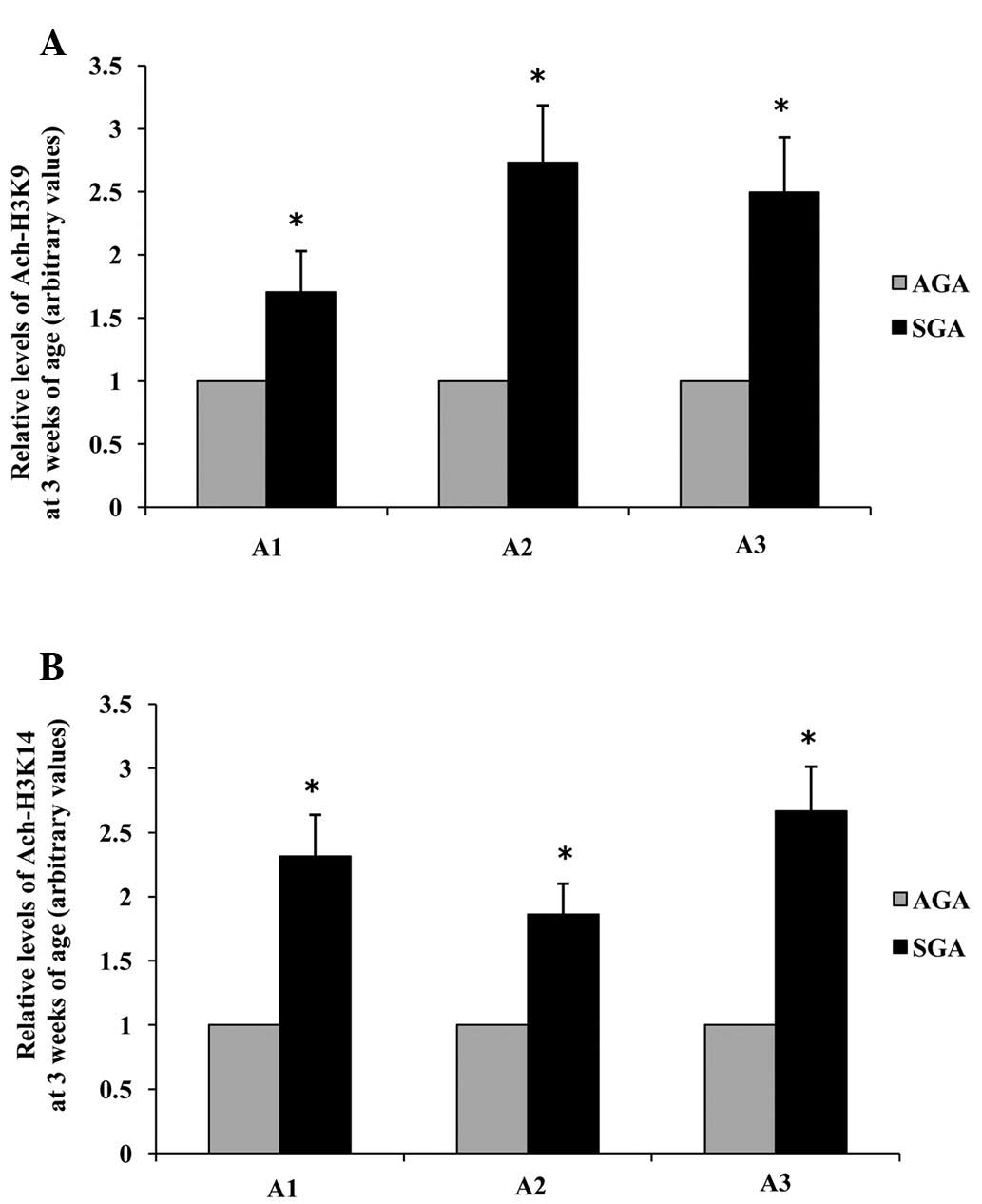 | Figure 5Effects of maternal undernutrition on
the epigenetic regulation of LPL promoter regions in rat livers at
3 weeks of age. (A) Acetylation of H3K9 and (B) H3K14. Quantitative
polymerase chain reaction analysis was performed using primers
specific to the proposed LPL element sites (A1, −87 to +177; A2,
−136 to −533; A3, −644 to −896). Data are presented as arbitrary
values, and are presented as the mean ± standard error of the mean.
*P<0.05, vs. the AGA rats. SGA, small for gestational
age; AGA, appropriate for gestational age; LPL, lipoprotein lipase;
Ach, acetylated; H3K, histone H3 lysine. |
Discussion
The present study demonstrated that 3-week-old SGA
male offspring exhibited increased hepatic TG levels. In liver
tissue, the fatty acid biosynthesis pathway facilitates excess
energy storage, either as cytosolic lipid droplets or circulating
TG-rich lipoproteins (21). These
TG may provide energy during times of deficiency following
oxidation; however, the excess accumulation of hepatic TG is a risk
factor for cardiovascular disease. Therefore, a better
understanding regarding the molecular determinants that control
fatty acid metabolism and hepatic TG levels may facilitate the
development of effective interventions that reduce the metabolic
risk factors for SGA male offspring.
The results of the present study demonstrated that
alterations in hepatic TG content were closely paralleled with
changes in hepatic mRNA and protein expression levels of LPL, which
implicated LPL in the development of hepatic lipid dysregulation. A
previous study reported that in fetal plasma from pregnancies
characterized by SGA, altered LPL expression appeared to be
associated with changes in FFA placental exchange, which may
contribute to an abnormal lipid profile (22). Kim et al (23) demonstrated that mice with
liver-specific LPL overexpression manifested hepatic steatosis and
insulin resistance. In addition, at the time of weaning, LPL mRNA
expression is nearly undetectable in normal rat livers; therefore,
the hepatic expression of LPL in SGA male rats may not only enable
the liver to hydrolyze TG from chylomicrons and very-low-density
lipoprotein, but may also lead to an increase in the hepatic uptake
of FFA, which may induce hepatic steatosis (24).
To the best of our knowledge, LPL mRNA expression
levels have been widely evaluated in the SGA placenta. Gauster
et al (25) compared the
LPL expression between normal pregnancies and those complicated
with SGA; the results demonstrated that LPL was markedly increased
(2.4-fold; P<0.015) in SGA placentas. In addition, Tabano et
al (26) detected an increase
in LPL mRNA expression in severe SGA cases with abnormal umbilical
blood flow, as compared with AGA placentas. The results of the
present study combined with findings from previous studies led us
to hypothesize that increased expression levels of LPL may persist
into postnatal life, and may be involved in the development of
metabolic disease in SGA male offspring.
It is well known that the regulation of LPL gene
expression is complex, occurring at transcriptional, translational
and post-translational levels. The present study hypothesized that
alterations in LPL expression in SGA rats may occur via sensitive
upstream transcriptional regulators, such as the LXR-α gene. To
further characterize the mechanism involved, ChIP coupled with qPCR
methods were used to study the exact mechanism. The results
suggested that LPL expression is mediated by LXR-α, which interacts
with LXRE sequences spanning the −3438 to −3423 promoter regions in
hepatic LPL. The nuclear receptor LXR-α is emerging as a key
regulator of lipid homeostasis, which is primarily expressed in the
liver, intestine, adipose tissue and macrophages. In addition to
LPL, LXR-α also regulates numerous genes involved in fatty acid
synthesis, including fatty acid synthase, acetyl CoA carboxylase
and the sterol-regulatory element binding protein 1 (27–29).
Previous animal studies have demonstrated that administration of
the LXR ligand, TO-901317, may cause severe fatty liver and obesity
(30,31). Since overexpression of the LXR-α
gene may increase fatty acid synthase via its target genes, it may
be considered a suitable target for therapeutic intervention, in
order to prevent hepatic fatty infiltration in SGA male
offspring.
The present study demonstrated that gene expression
is not the only molecular phenotype affected by maternal
nutritional manipulation. Epigenetic states can also be modified by
environmental factors, resulting in transcriptional expression or
silencing. Acetylation of H3K9 and H3K14 is generally believed to
be associated with actively transcribed genes (32,33),
which are congruent with the increased mRNA and protein expression
levels of LPL observed in the present study. Since hepatic
development occurs throughout neonatal and early postnatal life, it
is plausible that targeting this short period of development may
help reverse or prevent adverse hepatic epigenetic phenotypes in
SGA offspring.
In conclusion, the present study demonstrated that
maternal undernutrition during pregnancy and subsequent SGA may
result in postnatal alterations in the epigenetic characteristics
of the LPL gene, and increased binding of LXR-α to the LXRE in LPL
promoter regions. These alterations were associated with
predictable changes in LPL mRNA and protein expression levels, and
may result in elevated hepatic TG content.
Acknowledgments
The present study was supported by the National
Science Foundation of China (grant nos. 81170733 and 81000267).
References
|
1
|
Lees C, Marlow N, Arabin B, Bilardo CM,
Brezinka C, Derks JB, Duvekot J, Frusca T, Diemert A, Ferrazzi E,
et al TRUFFLE Group: Perinatal morbidity and mortality in
early-onset fetal growth restriction: Cohort outcomes of the trial
of randomized umbilical and fetal flow in Europe (TRUFFLE).
Ultrasound Obstet Gynecol. 42:400–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gluckman PD, Hanson MA, Cooper C and
Thornburg KL: Effect of in utero and early-life conditions on adult
health and disease. N Engl J Med. 359:61–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ross MG and Beall MH: Adult sequelae of
intrauterine growth restriction. Semin Perinatol. 32:213–218. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hediger ML, Overpeck MD, Kuczmarski RJ,
McGlynn A, Maurer KR and Davis WW: Muscularity and fatness of
infants and young children born small- or
large-for-gestational-age. Pediatrics. 102:E601998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ong KK, Ahmed ML, Emmett PM, Preece MA and
Dunger DB: Association between postnatal catch-up growth and
obesity in childhood: Prospective cohort study. BMJ. 320:967–971.
2000. View Article : Google Scholar
|
|
6
|
Law CM, Barker DJ, Osmond C, Fall CH and
Simmonds SJ: Early growth and abdominal fatness in adult life. J
Epidemiol Community Health. 46:184–186. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Parsons TJ, Power C and Manor O: Fetal and
early life growth and body mass index from birth to early adulthood
in 1958 British cohort: Longitudinal study. BMJ. 323:1331–1335.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ramasamy I: Recent advances in
physiological lipoprotein metabolism. Clin Chem Lab Med.
52:1695–1727. 2014. View Article : Google Scholar
|
|
9
|
Goldberg IJ and Merkel M: Lipoprotein
lipase: Physiology, biochemistry, andmolecular biology. Front
Biosci. 6:D388–D405. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Llobera M, Montes A and Herrera E:
Lipoprotein lipase activity in liver of the rat fetus. Biochem
Biophys Res Commun. 91:272–277. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Staels B and Auwerx J: Perturbation of
developmental gene expression in rat liver by fibric acid
derivatives: Lipoprotein lipase and alpha-fetoprotein as models.
Development. 115:1035–1043. 1992.PubMed/NCBI
|
|
12
|
Panadero M, Bocos C and Herrera E:
Relationship between lipoprotein lipase and peroxisome
proliferator-activated receptor-alpha expression in rat liver
during development. Physiol Biochem. 62:189–198. 2006. View Article : Google Scholar
|
|
13
|
Schoonjans K, Peinado-Onsurbe J, Lefebvre
AM, Heyman RA, Briggs M, Deeb S, Staels B and Auwerx J: PPARalpha
and PPARgamma activators direct a distinct tissue-specific
transcriptional response via a PPRE in the lipoprotein lipase gene.
EMBO. 15:5336–5348. 1996.
|
|
14
|
Dobrian AD, Lazar V, Sinescu C, Mincu D
and Simionescu M: Diabetic state induces lipid loading and altered
expression and secretion of lipoprotein lipase in human
monocyte-derived macrophages. Atherosclerosis. 153:191–201. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Astarita G, Taussiq MD, Bharadwaj
KG, DiPatrizio NV, Nave KA, Piomelli D, Goldberg IJ and Eckel RH:
Deficiency of lipoprotein lipase in neurons modifies the regulation
of energy balance and leads to obesity. Cell Metab. 13:105–113.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schoonjans K, Gelman L, Haby C, Briggs M
and Auwerx J: Induction of LPL gene expression by sterols is
mediated by a sterol regulatory element and is independent of the
presence of multiple E boxes. J Mol Biol. 304:323–334. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang Y, Repa JJ, Gauthier K and
Mangelsdorf DJ: Regulation of lipoprotein lipase by the oxysterol
receptors, LXRalpha and LXRbeta. J Biol Chem. 276:43018–43024.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sohi G, Marchand K, Revesz A, Arany E and
Hardy DB: Maternal protein restriction elevates cholesterol in
adult rat offspring due to repressive changes in histone
modification at the cholesterol 7alpha-hydroxlase promoter. Mol
Endocrinol. 25:785–798. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park JH, Stoffers DA, Nicholls RD and
Simmons RA: Development of type 2 diabetes following intrauterine
growth retardation in rats is associated with progressive
epigenetic silencing of Pdx1. J Clin Invest. 118:2316–2324.
2008.PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Jensen-Urstad AP and Semenkovich CF: Fatty
acid synthase and liver triglyceride metabolism: Housekeeper or
messenger? Biochim Biophys Acta. 1821:747–753. 2012. View Article : Google Scholar :
|
|
22
|
Cetin I, Giovannini N, Alvino G, Agostoni
C, Riva E, Giovannini M and Pardi G: Intrauterine growth
restriction is associated with changes in polyunsaturated fatty
acid fetal-maternal relationships. Pediatr Res. 52:750–755. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim JK, Fillmore JJ, Chen Y, Yu C, Moore
IK, Pypaert M, Lutz EP, Kako Y, Velez-Carrasco W, Goldberg IJ, et
al: Tissue-specific overexpression of lipoprotein lipase causes
tissue-specific insulin resistance. Proc Natl Acad Sci USA.
98:7522–75227. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pardina E, Baena-Fustegueras JA, Llamas R,
Catalán R, Galard R, Lecube A, Fort JM, Llobera M, Allende H,
Vargas V and Peinado-Onsurbe J: Lipoprotein lipase expression in
livers of morbidly obese patients could be responsible for liver
steatosis. Obes Surg. 19:608–616. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gauster M, Hiden U, Blaschitz A, Frank S,
Lang U, Alvino G, Cetin I, Desoye G and Wadsack C: Dysregulation of
placental endothelial lipase and lipoprotein lipase in intrauterine
growth-restricted pregnancies. J Clin Endocrinol Metab.
92:2256–2263. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tabano S, Alvino G, Antonazzo P, Grati FR,
Miozzo M and Cetin I: Placental LPL gene expression is increased in
severe intrauterine growth-restricted pregnancies. Pediatr Res.
59:250–253. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu H, Wu J, Yang M, Guo J, Zheng L, Peng
M, Zhang Q, Xiang Y, Cao J and Shen W: Involvement of liver X
receptor alpha in histone modifications across the target fatty
acid synthase gene. Lipids. 47:249–257. 2012. View Article : Google Scholar
|
|
28
|
Talukdar S and Hillgartner FB: The
mechanism mediating the activation of acetyl-coenzyme A
carboxylase-alpha gene transcription by the liver X receptor
agonist T0–901317. Lipid Res. 47:2451–2461. 2006. View Article : Google Scholar
|
|
29
|
Repa JJ, Liang G, Ou J, Bashmakov Y,
Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL and
Mangelsdorf DJ: Regulation of mouse sterol regulatory
element-binding protein-1c (SREBP-1c) by oxysterol receptors,
LXRalpha and LXRbeta. Genes Dev. 14:2819–2830. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cao G, Liang Y, Broderick CL, Oldham BA,
Beyer TP, Schmidt RJ, Zhang Y, Stayrook KR, Suen C, Otto KA, et al:
Antidiabetic action of a liver x receptor agonist mediated by
inhibition of hepatic gluconeogenesis. J Biol Chem. 278:1131–1136.
2003. View Article : Google Scholar
|
|
31
|
Chisholm JW, Hong J, Mills SA and Lawn RM:
The LXR ligand T0901317 induces severe lipogenesis in the db/db
diabetic mouse. J Lipid Res. 44:2039–2048. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim JM, Liu H, Tazaki M, Nagata M and Aoki
F: Changes in histone acetylation during mouse oocyte meiosis. J
Cell Biol. 62:37–46. 2003. View Article : Google Scholar
|
|
33
|
Schiltz RL, Mizzen CA, Vassilev A, Cook
RG, Allis CD and Nakatani Y: Overlapping but distinct patterns of
histone acetylation by the human coactivators p300 and PCAF within
nucleosomal substrates. J Biol Chem. 274:1189–1192. 1999.
View Article : Google Scholar : PubMed/NCBI
|