Introduction
Insulin resistance, defined as a process in which
the peripheral tissues, including the liver, muscle and fat, become
resistant to the actions of insulin, is the primary cause of type 2
diabetes (1–3). The liver has a central role in the
maintenance of plasma glucose homeostasis, and the induction of
hepatic insulin resistance has been reported to be important in its
involvement in the development of type 2 diabetes (4). In hepatocytes, decreased glucose
uptake and glycogen levels are the hallmark of insulin resistance
(5–7). In addition, insulin resistance occurs
at the molecular level through dysregulation in the complex network
of insulin signaling pathways (5,8).
3-Deoxyglucosone (3DG), a highly reactive dicarbonyl
product, has been identified as an intermediate product of the
Maillard reaction (9). 3DG has
been reported to be associated with diabetes and other age-related
human diseases (10–12). 3DG is elevated ~2-fold in plasma of
patients with diabetes, and is involved in the development of
diabetic complications (13–15).
In addition to exerting its potent ability to form advanced
glycation end-products, 3DG itself has certain biological
activities, including suppression of enzyme activity during glucose
metabolism and cell proliferation, induction of apoptosis and
inactivation of glutathione reductase (16–18).
Although evidence has indicated that methylglyoxal (MGO) induces
insulin resistance, and impairs insulin signaling in Sprague-Dawley
rats (19–21) and peripheral cells (22,23),
the concentration of MGO in a variety of foods, and the levels
produced from the Maillard reaction, are lower than 3DG. In
addition, Kiho et al (17)
reported that 3DG inhibits the activities of hexokinase and
glucose-6-phosphate dehydrogenase in crude extracts from the mouse
liver.
In our previous study, it was found that 3DG led to
impaired plasma glucose homeostasis in healthy mice (24) and, in non-diabetic seniors,
abnormal elevations in plasma 3DG were observed, which was
associated with impaired glucose regulation (25). Based on this evidence, it is
reasonable to hypothesize that 3DG may be important in the
development of insulin resistance. However, whether exposure to 3DG
induces insulin resistance and impairs insulin signaling pathways
directly in vitro remains be elucidated. To determine the
ability of 3DG to induce insulin resistance directly, the present
study determined the effect of exogenously added 3DG on glucose
uptake and the contents of glycogen in the hepatocellular
carcinoma, HepG2, cell line. The expression levels of insulin
signaling molecules involved in insulin resistance in the
hepatocytes were also investigated. The results may determine
whether the addition of exogenous 3DG can directly contribute to
inducing insulin resistance by impairing insulin signaling in HepG2
cells.
Materials and methods
Synthesis of 3DG
According to the method of Kato et al
(26), 3-DG was synthesized from
glucose. A hot solution of 6 g D-glucose, 3.3 g p-toluidine, 6.6 ml
acetic acid and 135 ml ethanol was stirred vigorously and heated in
an oil bath at 90°C for 30 min. Then, 9.9 g benzoylhydrazine was
added, followed by refluxing for 6–7 h. The reaction solution was
incubated at 4°C overnight, then the resulting precipitate
(3DG-bisbenzoylhydrazone) was collected by filtration through a
Buchner funnel (Aladdin Industrial Corporation, Shanghai, China),
and washed successively with 75 ml methanol and 75 ml diethyl ether
3 times. The product was then dried at room temperature. A solution
of the 3DG-bisbenzoylhydrazone product (3 g), 90 ml ethanol, 1.5 ml
acetic acid, 50 ml water and 1.6 ml freshly distilled benzaldehyde
(at 40–50 mmHg, 120–130°C) was refluxed at 90°C for 4 h. The
reaction mixture was incubated overnight at room temperature and
then the filtrate was collected through a Buchner funnel and then
concentrated to 70 ml using an RE-52 rotary evaporator (Shanghai
Yarong Biochemical Instrument Factory, Shanghai, China) and washed
6 times with 30 ml diethyl ether, then decolorized with 2 g
activated carbon. The concentrate was evaporated down to 3 ml and
10 ml of 95% ethanol was added. The solution was then charged on
Amberlite IR120B (H+ form) and Amberlite IR4B
(OH− form) ion-exchange resin columns. The final
solution was evaporated to a thick syrup, then the 3DG was purified
further using a flash column (silicagel 60;
chloroform/methanol/water ratio, 8.0:2.0:0.1). All chemical
reagents used were purchased from J&K Chemical, Ltd. (Shanghai,
China) without further purification. The product was identified
using infrared, 1H-nuclear magnetic resonance (NMR),
13C-NMR and mass spectroscopy (27).
HepG2 cell culture and treatment
The HepG2 hepatocellular carcinoma cells were
provided by the School of Biology and Basic Medical Sciences,
Soochow University (Suzhou, China), and were cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) containing 10% (v/v) fetal bovine serum
(Zhejiang Tianhang Biological Technology Co., Ltd., Huzhou, China)
and antibiotic solution (100 U/ml penicillin and 100 µg/ml
streptomycin; Beyotime Institute of Biotechnology, Shanghai, China)
at 37°C in a humidified atmosphere containing 5% CO2.
The cells were grown to 70–80% confluence and were seeded into
96/48-well plates at a density of 5×104 cells/well. The
cells were then either pre-treated with 3DG (10, 80 or 300 ng/ml)
in serum-free medium for 24 h, or remained untreated, with or
without subsequent exposure to insulin (2.0 or 6.6 IU/ml), for the
experimental groups.
Cell viability assay
Cell viability was measured using a standard
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT)
assay (Wuxi Zhanwang Chemical Co., Ltd., Wuxi, China). Briefly,
5×104 cells/well were seeded in 96-well microtiter
plates for 24 h at 37°C; the cells were then pre-incubated with or
without 3DG at final concentrations of 10, 50, 80, 300, 500, 800
and 1,000 ng/ml in serum-free medium supplemented with high
(H-DMEM; 25 mmol/l) or low (L-DMEM; 5.6 mmol/l) glucose (Gibco;
Thermo Fisher Scientific, Inc.) for 24 h at 37°C. The medium was
subsequently removed, and 200 µl of MTT was added to a final
concentration of 0.5 mg/ml. After 4 h, 150 µl dimethyl
sulfoxide solution (Wuxi Zhanwang Chemical Reagent Co., Ltd., Wuxi,
China) was added to solubilise the MTT formazan. The plates were
placed on a mechanical shaker (Shanghai Centrifuge Institute Co.,
Ltd., Shanghai, China) for 10 min at room temperature, and the
optical density (OD) was read at 490 nm using an enzyme-linked
immunometric meter SpectraMax M2 (Molecular Devices, LLC,
Sunnyvale, CA, USA), as previously described (28).
Measurement of glucose uptake
The glucose uptake was measured using
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)
amino]-2-deoxyglucose (2-NBDG; Cayman Chemical Co., Ann Arbor, MI,
USA), comparable to the study by Engelbrecht et al (29). Following treatment of the cells
(5×107 cells/well) with or without different
concentrations of 3DG in L-DMEM for 24 h, the cells were washed
with Krebs-ringer bicarbonate (KRb) buffer (4A Biotech Co. Ltd.,
Beijing, China) comprising 129 mM NaCl, 4.7 mM KCl, 1.2 mM
KH2PO4, 1.0 mM CaCl2, 1.2 mM
MgSO4, 5.0 mM NaHCO3 and 10.0 mM HEPES. In
the insulin-treated group, the cells were then exposed to 3.60
IU/ml insulin, and 100 µl serum-free KRb buffer
(supplemented with 160 µM 2-NBDG) was added to the medium
and incubated for 30 min at 37°C. The cells were washed twice with
KRb buffer and the radioactivity incorporated into the cells was
measured using a fluorescence microplate reader
(excitation/emission, 488/520; Model 680, Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Subsequently, the medium was replaced
with L-DMEM supplemented with 200 µl MTT, and continued to
culture. After 4 h incubation at 37°C, the OD at 490 nm was
measured using a microplate reader. The error was corrected using
an MTT assay.
Measurement of glycogen content
The glycogen levels in the HepG2 cells were
determined using a glycogen assay kit in the presence or absence of
3.6 IU/ml insulin. Briefly, following 3DG treatment of the cells
for 24 h, the medium containing L-DMEM was removed and the cells
were washed twice with ice-cold phosphate-buffered saline (PBS;
Beyotime Institute of Biotechnology) and incubated with serum-free
L-DMEM and insulin for 24 h. Subsequently, glycogen in the cells
was extracted using 66% (v/v) ethanol and centrifuged for 10 min at
8,000 × g at 4°C to remove the supernatant. The precipitates of
each sample were mixed with 0.5 ml water, and the medium was
subsequently added into 1 ml 0.2% (w/v) anthrone reagents (Rsbio,
Shanghai, China). The tubes were boiled for 30 min. Absorbance at
620 nm was measured using an enzyme-linked immunometric meter. A
standard glycogen curve (50, 25, 12.5, 6.25, 3.12 and 1.60 mg/ml)
was calculated using the above method.
Western blot analysis
The HepG2 cells were rendered insulin resistant by
treatment with 3DG, as described above. The medium containing
L-DMEM and 3DG was removed from the cells, and the cells were then
washed twice with ice-cold PBS and solubilised in IP lysin buffer
containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, 2 mM
SDS, 25 mM β-glycerophoaphate, 1 mM EDTA, 1 mM
Na3VO4 and 0.5 µg/ml leupeptin
(Beyotime Institute of Biotechnology). Following centrifugation at
12,000 × g at 4°C for 20 min, the supernatants were collected and
used for Western blot analysis. The total protein concentrations
were determined using a bicinchoninic acid protein (BCA) assay kit
(cat. no. P0012; Beyotime Institute of Biotechnology) comprised of
BCA kit A (cat. no P0012-1), BCA kit B (cat. no. P0012-2) and
standard proteins (cat. no. P0012-3). The proteins (80–120
µg) were loaded onto a 12% SDS-polyacrylamide gel (Thermo
Fisher Scientific, Inc.), then subjected to electrophoresis and
transferred onto polyvinylidene fluoride membranes (Merck
Millipore, Darmstadt, Germany). The membranes were blocked for 1 h
in Tris-buffered saline with 1% Tween (TBST; Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China) containing 5%
dry milk. The membranes were washed with PBS containing 0.05%
Tween-20 three times, and incubated at 4°C overnight with the
following antibodies (dilution, 1:1,000): Rabbit polyclonal
anti-glucose transporter 2 (GLUT-2; cat. no. ab54460; Abcam,
Cambridge, UK); and rabbit monoclonal anti-glycogen synthase
kinase-3 (GSK-3)a/β (cat. no. 5676), rabbit polyclonal
anti-p-insulin receptor substrate 1 (IRS-1; cat. no. 3070), rabbit
polyclonal anti-phosphoinositide 3-kinase (PI3K)-p85 (cat. no.
4292), rabbit polyclonal anti-PI3K-p110 (cat. no. 4255) and rabbit
polyclonal anti-AKT (cat. no. 9272), all purchased from Cell
Signaling Technology, Inc., Danvers MA, USA. Following washing 4
times for 5 min each in TBST, the membranes were incubated with
goat anti-rabbit secondary antibody [1:1,000; cat. no. GAR0072;
Multi Sciences (Lianke) Biotech Co., Ltd., Hangzhou, China] for 2 h
and visualized using an ECL detection kit (Beijing Solarbio Science
& Technology Co., Ltd.). Quantification of protein bands was
performed using Image-J software (version 1.42).
Statistical analysis
The results of the experiments are expressed as the
mean ± standard deviation. SPSS software (version 14.0; SPSS, Inc.,
Chicago, IL, USA) was used for statistical analysis The statistical
significance of differences were analyzed using Student's
t-test. P≤0.05 was considered to indicate a statistically
significant difference. In the following, n describes the number of
repetitions of independent experiments.
Results
3DG does not alter HepG2 cell viability
at concentrations between 10 and 300 ng/ml
The HepG2 cells were exposed to different
concentrations of 3DG supplemented with H-DMEM or L-DMEM for 24 h
to examine the effects of 3DG on cell viability. Different
concentrations of 3DG were assessed, ranging between 10 and 1,000
ng/ml, and cell viability was measured using an MTT assay. As shown
in Fig. 1, compared with the
untreated control, 3DG did not alter HepG2 cell viability at
concentrations between 10 and 300 ng/ml, whereas 3DG exhibited a
degree of inhibitory activity against the growth of the HepG2 cells
at concentrations of 500 or 1,000 ng/ml.
Non-cytotoxic concentrations of 3DG
induce decreased 2-NBDG uptake and glycogen content in
insulin-stimulated HepG2 cells
To determine whether 3DG leads to hepatic insulin
resistance, the uptake of 2-NBDG and glycogen content in the HepG2
cells were measured following treatment with non-cytotoxic
concentrations (10–300 ng/ml) of 3DG for 24 h. Compared with the
untreated control cells, 2-NBDG uptake was significantly increased
following insulin stimulation (112.61±8.42, vs. 198.85±15.65;
P<0.001). The uptake of 2-NBDG remained unchanged in the
3DG-only treated cells, however, the cells co-incubated with 80
ng/ml (162.93±20.49; P<0.05) or 300 ng/ml (151.44±11.79;
P<0.01) 3DG and insulin showed significant dose-dependent
decreases in 2-NBDG uptake, compared with the cells exposed to
insulin only (198.85±15.65; Fig.
2A). As shown in Fig. 2B,
exposure to insulin significantly increased glycogen content,
compared with the control (29.743±3.712, vs. 10.151±1.102
µmol/mg, respectively). The cells treated with different
concentrations of 3DG showed significant dose-dependent decreases
in glycogen content, at concentrations of 80 ng/ml (22.060±1.821;
P<0.05) and 300 ng/ml (16.568±1.200 µmol/mg; P<0.01),
compared with the cells exposed to insulin alone (29.743±3.712
µmol/mg) However, the glycogen content remained unchanged in
the 3DG-only treated cells, compared with the normal group. The
effect of 3DG treatment became significant from a concentration of
80 ng/ml. Lower concentrations of 3DG did not significantly induce
decreased glucose uptake or glycogen content.
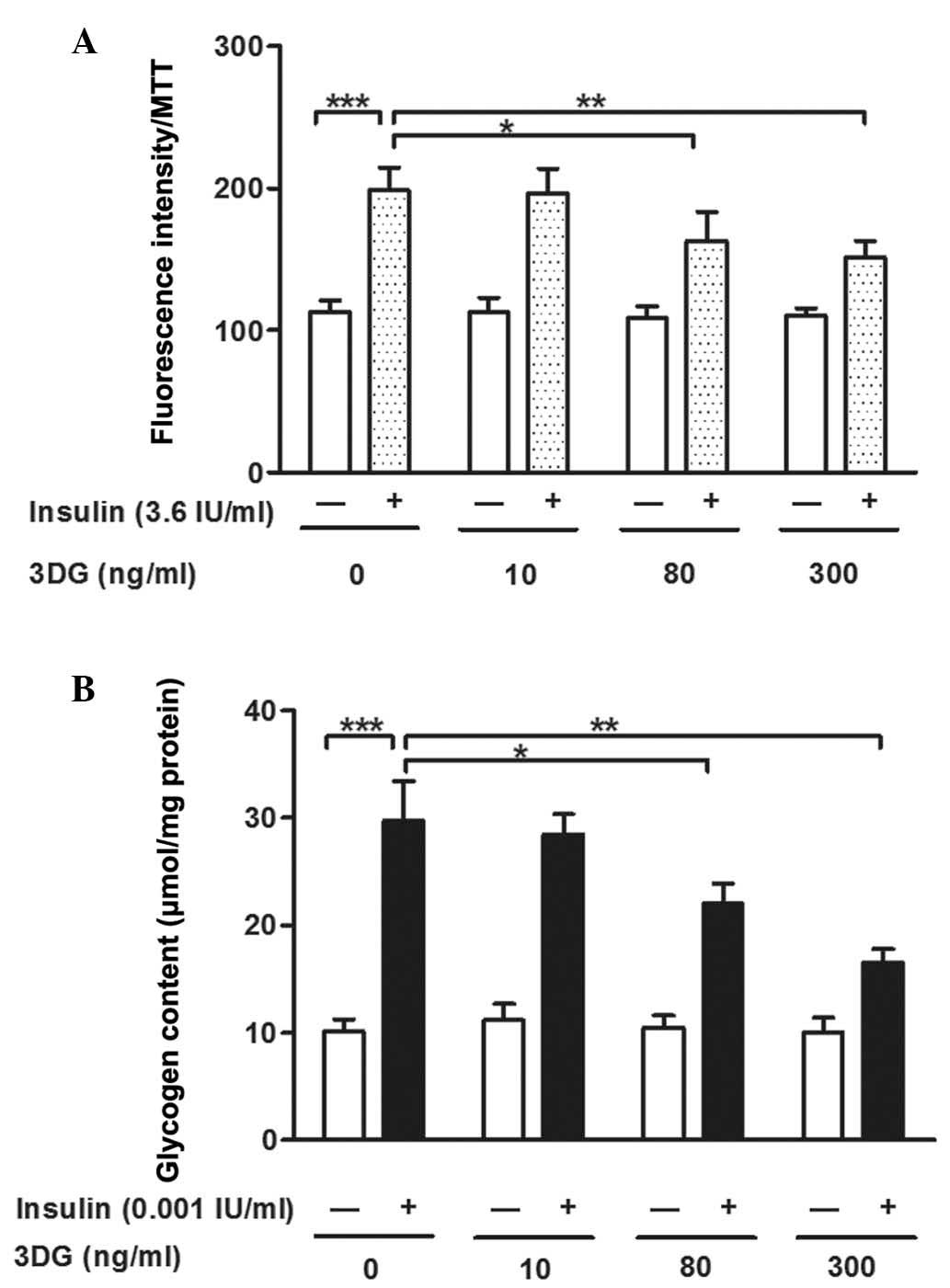 | Figure 2Effects of treatment with
non-cytotoxic concentrations of 3DG, with and without insulin
stimulation, on 2-NBDG uptake and glycogen content in HepG2 cells.
(A) HepG2 cells were stained with 2-NBDG for 30 min following
treatment with exogenous 3DG (10, 80 and 300 ng/ml, 24 h).
Fluorescence intensity was measured on a fluorescence microplate
reader, and an MTT assay was used to correct error (n=5).
***P<0.001, vs. untreated cells;
*P<0.05 and **P<0.01, vs. untreated
cells with insulin. (B) Glycogen content in the HepG2 cells was
determined using a glycogen assay kit following treatment with
exogenous 3DG (10, 80 and 300 ng/ml) for 24 h (n=5).
***P<0.001, vs. untreated cells;
*P<0.05 and **P<0.01, vs. untreated
cells with insulin. Data are presented as the mean ± standard
deviation. 3DG, 3-Deoxyglucosone; 2-NBDG
2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxyglucose;
MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium
bromide. |
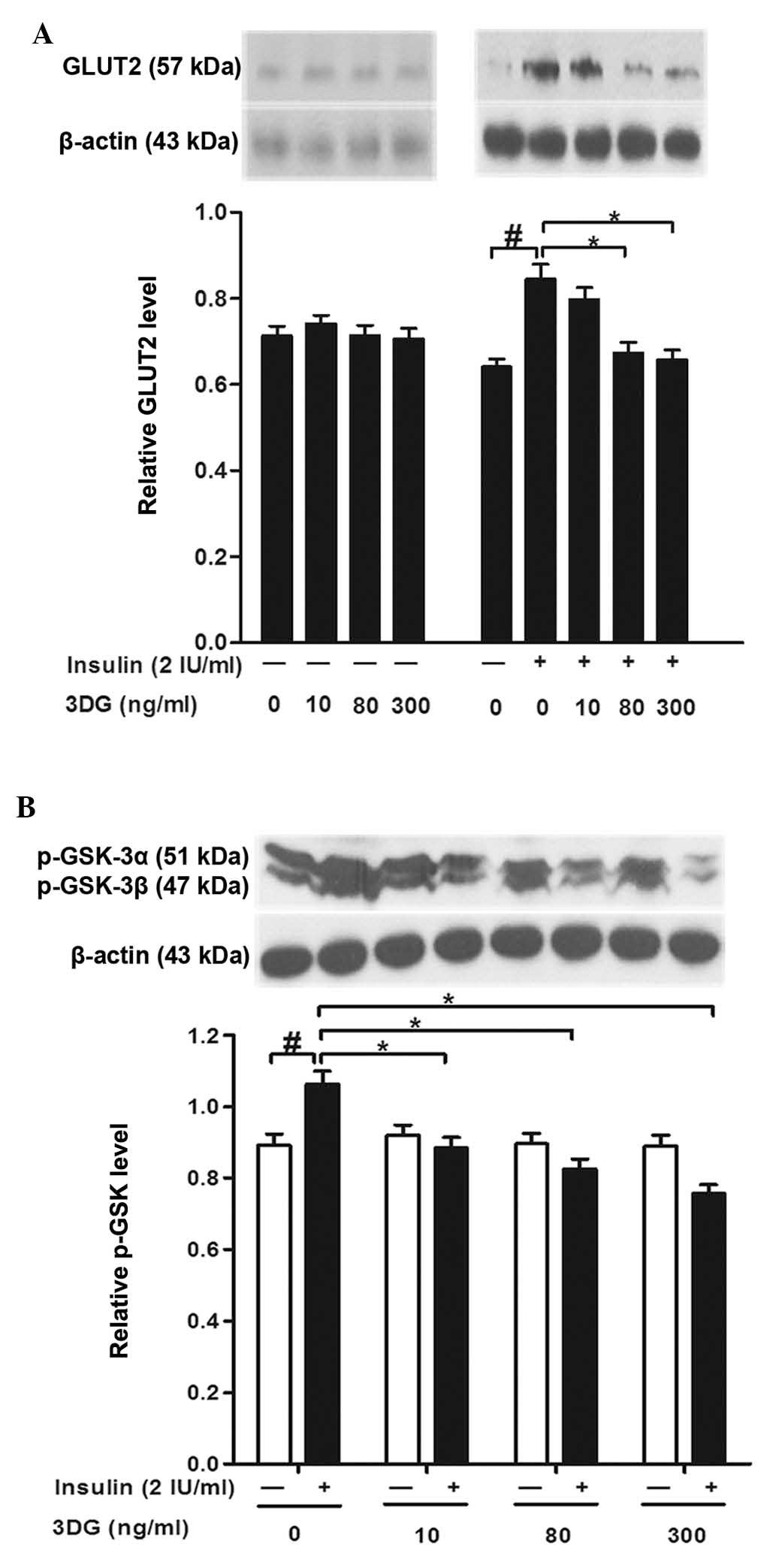
Treatment with non-cytotoxic
concentrations of 3DG decreases the insulin-induced expression of
GLUT2 and p-GSK-3a/β in HepG2 cells
To further elucidate the action of 3DG, the present
study evaluated the expression profile of proteins, which are
important for glucose uptake and glycogen content in HepG2 cells.
In the present study, treatment of the HepG2 cells with insulin
alone induced a significant increase in the expression levels of
GLUT2 and p-GSK-3a/β, compared with the control. Although the
expression levels of GLUT2 and p-GSK-3a/β were not altered by
non-cytotoxic concentrations of 3DG in the 3DG-only treated celss,
significant decreases in the protein expression levels of GLUT2 and
p-GSK-3a/β were observed following exposure of the cells to 3DG at
concentrations of 80 and 300 ng/ml for 24 h with insulin
stimulation (Fig. 3).
Effects of non-cytotoxic concentrations
of 3DG on insulin signaling in HepG2 cells: Expression and
phosphorylation of IR-β, IRS-1, PI3K-p85, PI3K-p110 and AKT
Glucose uptake and glycogen synthesis in the liver
are regulated via the activation of IRS-1, PI3K and AKT (30). Exogenous 3DG treatment had no
significant effect on the expression levels of IR-β and IRS-1,
however, treatment of the cells with 300 ng/ml 3DG reduced the
insulin-stimulated tyrosine phosphorylation of IRS-1 (Fig. 4A). To investigate the consequences
of the reduced tyrosine phosphorylation of IRS-1, the expression
levels of the downstream target proteins, PI3K-p85, PI3K-p110, AKT
and p-AKT, were determined. As shown in Fig. 4A, 3DG treatment induced significant
decreases in the protein expression levels of the PI3K p85 and p110
subunits. Furthermore, 3DG led to a dose-dependent decrease in the
phosphorylation of AKT, which was not accompanied by changes in the
level of AKT (Fig. 4B).
Discussion
Insulin is important in the stimulation of glucose
uptake, the impairment of which is a major factor responsible for
insulin resistance. In additional, insulin acts by decreasing
glucose production and enhancing its storage as glycogen in
hepatocytes, the disorder of which is also considered to be an
underlying mechanism of insulin resistance (6,7,31).
In the present study, it was observed that exposure of the HepG2
cells to 3DG for 24 h at concentrations of 80 and 300 ng/ml
decreased glucose uptake and glycogen content with insulin
stimulation, but had no effect on glucose uptake or glycogen
content in the absence of insulin (Fig. 2). In addition, decreased glucose
consumption was observed in the 3DG-treated HepG2 cells following
stimulation with insulin (data not shown). In agreement with a
study by Kiho et al (17),
these results suggested the association between 3DG and the
development of insulin resistance.
In hepatocytes, the primary mechanism of
insulin-stimulated glucose uptake and release in the fed and fasted
states, respectively, is through GLUT2, which has a low apparent
affinity for glucose (Km ~17 mmol/l) and is expressed at
high levels in the liver (32,33).
The high Km value for glucose indicates that the rate of
glucose transport by GLUT2 varies with the concentrations of
glucose under physiological conditions. In the present study, the
expression level of GLUT2 was reduced significantly following
exposure of the HepG2 cells to 3DG at 80 and 300 ng/ml for 24 h
with insulin stimulation. However, in the absence of insulin, no
significant decrease in the expression level of GLUT2 was observed
(Fig. 3A). However, the precise
role of GLUT2 in glucose transport in the liver remains to be fully
elucidated. Another glucose transporter isoform, GLUT1, which shows
low levels of expression in hepatocytes, has been identified to
collaborate with GLUT2 to mediate glucose uptake (34). In the present study, it was shown
that the protein expression of GLUT1 was reduced following
stimulation with insulin and treatment of the HepG2 cells with 3DG
(data not shown). Furthermore, the effect of 3DG on GLUT2 in fed
and fasted states in animal models is under investigation. GSK-3 is
a serine/threonine kinase, which is associated with several
diseases, including cancer, diabetes, inflammation, and Alzheimer's
disease (35,36). It has been reported that GSK-3
inactivation is involved in the positive regulation of
hepatocellular carcinoma cell proliferation (37,38).
In addition, insulin phosphorylates and inactivates GSK-3, leading
to the activation of the glycogen synthase (GS), which is
responsible for the storage of glucose in the liver (39). Therefore, the activation of GSK-3
promotes the phosphorylation and inactivation of GS, thereby
decreasing glycogen synthesis. The findings in the HepG2 cells in
the present study were consistent with the above. Although the
expression level of p-GSK-3 was not altered by 3DG treatment under
basal conditions, a reduction in the phosphorylation of GSK-3 was
observed in the 3DG-treated cells stimulated with insulin (Fig. 3B). In the 3DG-only treated cells,
the HepG2 cell viability was inhibited at concentrations of 500 and
1,000 ng/ml (Fig. 1), possibly due
to mechanisms including the decreased expression of GLUT1 (40) and phosphorylation of GSK-3. In
addition, it has been shown that 3DG can induce the apoptosis of
U937 monocytic leukemia cells (41), and the results of the present study
indicated that 3DG directly impaired insulin action in the HepG2
cells.
Insulin signaling pathways, for example the
IR/IRS/PI3K/AKT signaling pathway, are important in regulating the
expression of GLUT2 (42–44) and GSK-3 (7,45,46).
When insulin binds to IR, the intrinsic tyrosine kinase activity of
IR is activated. The activated receptor then phosphorylates its own
receptor and IRS, thereby dissociating IRS from IR to bind the PI3K
regulatory subunit, p85, following which the p110 subunit is
activated. Activated PI3K produces phosphatidylinosito (3,4,5)-triphosphate via the phosphorylation of
phosphatidylinosito (4,5)-bisphosphate, and activates AKT through
binding. The activated AKT inactivates GSK-3 at
ser9/21 and activates GLUT2 to enhance
glucose uptake (47). To clarify
the effect of 3DG on insulin signaling, the present study directly
treated HepG2 cells with 3DG. Reductions in the phosphorylation of
IRS-1 and AKT, and in the expression levels of PI3K-p110 and
PI3K-p85 were observed (Fig. 4).
These results indicated that treatment of the cells with 3DG may
have induced significant decreases in the protein expression levels
of GLUT2 and p-GSK-3 by impairing the insulin signaling
pathway.
3DG, a reactive 1,2-dicarbonyl compound, is formed
non-enzymaticallly in the course of the Maillard reaction and in
the caramelization processes in food (12,48).
The contents of 3DG in foods, including balsamic vinegar, honey and
bakery products, have been published (49,50).
In addition to the Maillard reaction, 3DG is also synthesized via
fruc-toseamine-3-kinase (51) and
the polyol pathway (52) in
vivo. In the present study, it was observed that the direct
addition of exogenous 3DG contributes to insulin resistance in
HepG2 cells. These results suggested that 3DG may be involved in
inducing impaired glucose regulation and worsening of the diabetic
condition as the plasma concentration of 3-DG is elevated.
Therefore, 3DG may offer potential as a novel target for the
prevention and therapy of diabetes and its complications.
In conclusion, the present study demonstrated for
the first time, to the best of our knowledge, that exogenous 3DG
impaired insulin signaling, which may have led to decreased
insulin-stimulated glucose uptake and glycogen synthesis, and
contributed to insulin resistance in the HepG2 cells. These
findings, in combination with those of previous studies (24,25)
indicated that 3DG is an independent factor contributing to insulin
resistance. Further elucidation of this novel 3DG-mediated
mechanism may assist in establishing novel and more effective
preventative strategies to improve prevention and treatment in
patients with insulin resistance and type 2 diabetes. Reducing the
ingestion and production of exogenous or endogenous 3DG,
respectively, may be applied for the clinical management of
diabetes due to 3DG being a potential risk factor for the
progression of diabetes.
Acknowledgments
This study was supported by research funds from the
Suzhou Science and Technology Department (grant nos. SYS201423 and
SYS201153) and the Suzhou Youth Science and Education Project
(grant no. KJXW2014027).
References
|
1
|
Petersen KF and Shulman GI: New insights
into the pathogenesis of insulin resistance in humans using
magnetic resonance spectroscopy. Obesity (Silver Spring). 14(Suppl
1): S34–S40. 2006. View Article : Google Scholar
|
|
2
|
Gallagher EJ, LeRoith D and Karnieli E:
The metabolic syndrome-from insulin resistance to obesity and
diabetes. Endocrinol Metab Clin North Am. 37:559–579. 2008.
View Article : Google Scholar
|
|
3
|
Goldstein BJ: Insulin resistance as the
core defect in type 2 diabetes mellitus. Am J Cardiol. 90:3G–10G.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taniguchi CM, Ueki K and Kahn R:
Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of
metabolism. J Clin Invest. 115:718–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hunter SJ and Garvey WT: Insulin action
and insulin resistance: Diseases involving defects in insulin
receptors, signal transduction and the glucose transport effector
system. Am J Med. 105:331–345. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leclercq IA, Da Silva Morais A, Schroyen
B, Van Hul N and Geerts A: Insulin resistance in hepatocytes and
sinusoidal liver cells: Mechanisms and consequences. J Hepatol.
47:142–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dou L, Zhao T, Wang L, Huang X, Jiao J,
Gao D, Zhang H, Shen T, Man Y, Wang S and Li J: miR-200s contribute
to interleukin-6 (IL-6)-induced insulin resistance in hepatocytes.
J Biol Chem. 288:22596–22606. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sechi LA and Bartoli E: Mechanisms of
insulin resistance leading to hypertension: What we can learn from
experimental models. J Investig Med. 45:238–251. 1997.PubMed/NCBI
|
|
9
|
Shin DB, Hayase F and Kato H:
Polymerization of proteins caused by reaction with sugars and the
formation of 3-deoxyglucosone under physiological conditions. Agric
Biol Chem. 52:1451–1458. 1988. View Article : Google Scholar
|
|
10
|
Kusunoki H, Miyata S, Ohara T, Liu BF,
Uriuhara A, Kojima H, Suzuki K, Miyazaki H, Yamashita Y, Inaba K
and Kasuga M: Relation between serum 3-deoxyglucosone and
development of diabetic microangiopathy. Diabetes Care.
26:1889–1894. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Supplementto American Academy of
Dermatology 66th Annual Meeting: Inhibitors of 3-deoxyglucosone
(3DG) treat aging and inflamed skin by preventing the form action
of AGEs, oxidative stress and free radicals. San Antonio, Texas. J
Am Acad Dermatol. 2312008.
|
|
12
|
Niwa T: 3-Deoxyglucosone: Metabolism,
analysis, biological activity and clinical implication. J
Chromatogr B Biomed Sci Appl. 731:23–36. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lal S, Kappler F, Walker M, Orchard TJ,
Beisswenger PJ, Szwergold BS and Brown TR: Quantitation of
3-deoxyglucosone levels in human plasma. Arch Biochem and Biophys.
342:254–260. 1997. View Article : Google Scholar
|
|
14
|
Hamada Y, Nakamura J, Fujisawa H, Yago H,
Nakashima E, Koh N and Hotta N: Effects of glycemic control on
plasma 3-deoxyglucosone levels in NIDDM patients. Diabetes Care.
20:1466–1469. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Beisswenger PJ, Howell SK, O'Dell RM, Wood
ME, Touchette AD and Szwergold BS: alpha-Dicarbonyls increase in
the postprandial period and reflect the degree of hyperglycemia.
Diabetes Care. 24:726–732. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sakiyama H, Takahashi M, Yamamoto T,
Teshima T, Lee SH, Miyamoto Y, Misonou Y and Taniguchi N: The
internalization and metabolism of 3-deoxyglucosone in human
umbilical vein endothelial cells. J Biochem. 139:245–253. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kiho T, Asahi T, Usui S, Matsunaga T and
Ukai S: Effect of 3-deoxyglucosone on the activities of enzymes
responsible for glucose metabolism in mouse liver. Biol Pharm Bull.
19:1106–1108. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sassi-Gaha S, Loughlin DT, Kappler F,
Schwartz ML, Su B, Tobia AM and Artlett CM: Two dicarbonyl
compounds, 3-deoxyglucosone and methylglyoxal, differentially
modulate dermal fibroblasts. Matrix Biol. 29:127–134. 2010.
View Article : Google Scholar
|
|
19
|
Dhar A, Desai KM and Wu L: Alagebrium
attenuates acute methylglyoxal-induced glucose intolerance in
Sprague-Dawley rats. Br J Pharmacol. 159:166–175. 2010. View Article : Google Scholar :
|
|
20
|
Jia X and Wu L: Accumulation of endogenous
methylglyoxal impaired insulin signaling in adipose tissue of
fructose-fed rats. Mol Cell Biochem. 306:133–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhar A, Dhar I, Jiang B, Desai KM and Wu
L: Chronic methylglyoxal infusion by minipump causes pancreatic
beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley
rats. Diabetes. 60:899–908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Riboulet-Chavey A, Pierron A, Durand I,
Murdaca J, Giudicelli J and Van Obberghen E: Methylglyoxal impairs
the insulin signaling pathways independently of the formation of
intracellular reactive oxygen species. Diabetes. 55:1289–1299.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fiory F, Lombardi A, Miele C, Giudicelli
J, Beguinot F and Van Obberghen E: Methylglyoxal impairs insulin
signalling and insulin action on glucose-induced insulin secretion
in the pancreatic beta cell line INS-1E. Diabetologia.
54:2941–2952. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Jiang Gr and Zhang Lr: Effects of
3-deoxyglucosone on blood glucose of normal mice. Chin J Diabetes.
18:220–222. 2010.
|
|
25
|
Jiang G, Zhang L, Ji Q, Wang F, Xu H,
Huang F and Wang C: Accumulation of plasma 3-deoxyglucosone
impaired glucose regulation in Chinese seniors: Implication for
senile diabetes? Diabetes Metab Syndr. 6:140–145. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kato H, van Chuyen N, Shinoda T, Sekiya F
and Hayase F: Metabolism of 3-deoxyglucosone, an intermediate
compound in the Maillard reaction, administered orally or
intravenously to rats. Biochim Biophys Acta. 1035:71–76. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang GL, Zhang LV, Wang F, Xu H and Fei
D: Synthesis and structure analysis of the 3-deoxyglucosone (3-DG).
J Soochow Univ. 27:60–68. 2011.
|
|
28
|
Zhang L, Jiang G, Yao F, He Y, Liang G,
Zhang Y, Hu B, Wu Y, Li Y and Liu H: Growth inhibition and
apoptosis induced by osthole, a natural coumarin, in hepatocellular
carcinoma. PLoS One. 7:e378652012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Engelbrecht B, Mattern Y, Scheibler S,
Tschoepe D, Gawlowski T and Stratmann B: Methylglyoxal impairs
GLUT4 trafficking and leads to increased glucose uptake in L6
myoblasts. Horm Metab Res. 46:77–84. 2014.
|
|
30
|
Cordero-Herrera I, Martín MÁ, Goya L and
Ramos S: Cocoa flavonoids attenuate high glucose-induced insulin
signalling blockade and modulate glucose uptake and production in
human HepG2 cells. Food Chem Toxicol. 64:10–19. 2014. View Article : Google Scholar
|
|
31
|
Schinner S, Scherbaum WA, Bornstein SR and
Barthel A: Molecular mechanisms of insulin resistance. Diabetic
Med. 22:674–682. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mueckler M and Thorens B: The SLC2 (GLUT)
family of membrane transporters. Mol Aspects Med. 34:121–138. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hosokawa M and Thorens B: Glucose release
from GLUT2-null hepatocytes: Characterization of a major and a
minor pathway. Am J Physiol Endocrinol Metab. 282:E794–E801. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Levitsky LL, Zheng Q, Mink K and Rhoads
DB: GLUT-1 and GLUT-2 mRNA, protein, and glucose transporter
activity in cultured fetal and adult hepatocytes. Am J Physiol.
267:E88–E94. 1994.PubMed/NCBI
|
|
35
|
Martinez A, Castro A, Dorronsoro I and
Alonso M: Glycogen synthase kinase 3 (GSK-3) inhibitors as new
promising drugs for diabetes, neurodegeneration, cancer and
inflammation. Med Res Rev. 22:373–384. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Medina M and Castro A: Glycogen synthase
kinase-3 (GSK-3) inhibitors reach the clinic. Curr Opin Drug Discov
Devel. 11:533–543. 2008.PubMed/NCBI
|
|
37
|
Ban KC, Singh H, Krishnan R and Seow HF:
GSK-3beta phosphorylation and alteration of beta-catenin in
hepatocellular carcinoma. Cancer Lett. 199:201–208. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ibrahim SH, Akazawa Y, Cazanave SC, Bronk
SF, Elmi NA, Werneburg NW, Billadeau DD and Gores GJ: Glycogen
synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte
lipoapoptosis. J Hepatol. 54:765–772. 2011. View Article : Google Scholar :
|
|
39
|
Nachar A, Vallerand D, Musallam L, Lavoie
L, Badawi A, Arnason J and Haddad PS: The action of antidiabetic
plants of the canadian james bay cree traditional pharmacopeia on
key enzymes of hepatic glucose homeostasis. Evid Based Complement
Alternat Med. 2013:1898192013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xu YY, Wu TT, Zhou SH, Bao YY, Wang QW,
Fan J and Huang YP: Apigenin suppresses GLUT-1 and p-AKT expression
to enhance the chemosensitivity to cisplatin of laryngeal carcinoma
Hep-2 cells: An in vitro study. Int J Clin Exp Pathol. 7:3938–3947.
2014.PubMed/NCBI
|
|
41
|
Okado A, Kawasaki Y, Hasuike Y, Takahashi
M, Teshima T, Fujii J and Taniguchi N: Induction of apoptotic cell
death by methylglyoxal and 3-deoxyglucosone in macrophage-derived
cell lines. Biochem Biophys Res Commun. 225:219–224. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Song Z, Wang H, Zhu L, Han M, Gao Y, Du Y
and Wen Y: Curcumin improves high glucose-induced INS-1 cell
insulin resistance via activation of insulin signaling. Food
Function. 6:461–469. 2015. View Article : Google Scholar
|
|
43
|
Ruan CT, Lam SH, Chi TC, Lee SS and Su MJ:
Borapetoside C from Tinospora crispa improves insulin sensitivity
in diabetic mice. Phytomedicine. 19:719–724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Manna P and Jain SK: Decreased hepatic
phosphatidylinositol-3,4,5-triphosphate (PIP3) levels and impaired
glucose homeostasis in type 1 and type 2 diabetic rats. Cell
Physiol Biochem. 30:1363–1370. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang ZB, Zeng HC, Wei HS, Yi GH, Yu J,
Wang YT, Zhang YL and Yin WD: NO-1886 ameliorates glycogen
metabolism in insulin-resistant HepG2 cells by GSK-3β signalling. J
Pharm Pharmacol. 64:293–301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lavoie L, Band CJ, Kong M, Bergeron JJ and
Posner BI: Regulation of glycogen synthase in rat hepatocytes.
Evidence for multiple signaling pathways. J Biol Chem.
274:28279–28285. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Saltiel AR and Kahn CR: Insulin signalling
and the regulation of glucose and lipid metabolism. Nature.
414:799–806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kroh LW: Caramelisation in food and
beverages. Food Chem. 51:373–379. 1994. View Article : Google Scholar
|
|
49
|
Hellwig M, Degen J and Henle T:
3-deoxygalactosone, a 'new' 1,2-dicarbonyl compound in milk
products. J Agric Food Chem. 58:10752–10760. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Degen J, Hellwig M and Henle T:
1,2-dicarbonyl compounds in commonly consumed foods. J Agric Food
Chem. 60:7071–7079. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Brown TR, Su B, Brown KA, Schwartz MA,
Tobia AM and Kappler F: Modulation of in vivo 3-deoxyglucosone
levels. Biochem Soc Trans. 31:1433–1437. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lal S, Szwergold BS, Taylor AH, Randall
WC, Kappler F, Wells-Knecht K, Baynes JW and Brown TR: Metabolism
of fructose-3-phosphate in the diabetic rat lens. Arch Biochem
Biophys. 318:191–199. 1995. View Article : Google Scholar : PubMed/NCBI
|