Introduction
Lung complications, such as pneumonitis and
fibrosis, frequently occur in the thoracic primary and metastatic
tumors following thoracic radiotherapy (RT) (1,2) and
sets a limit to its use and dosage. However, the underlying
molecular mechanisms of radiation-induced pulmonary fibrosis (RIPF)
have remained elusive. Inflammatory cytokines, transforming growth
factor (TGF)-β and chronic reactive oxygen species (ROS) are known
to contribute to RIPF. ROS generated in large quantities by
irradiation-induced cellular damage and involved in various
signaling pathways and DNA fragmentation, which induces apoptosis
in the initial phase of tissue damage (3–5). In
addition, radiation-induced late normal tissue injury, including
RIPF, is thought to be caused by chronic oxidative stress and
inflammation. Therefore, anti-oxidant enzymes have been suggested
to ameliorate radiation-induced chronic injury to normal tissue
(6–8).
Nicotinamide adenine dinucleotide phosphate (NADPH)
oxidases (NOXs) generate ROS (9)
and catalyze the transfer of electrons from cytosolic NADPH to
molecular O2 via the membrane-bound catalytic NOX or
dual oxidase sub-units (9). NOXs
are implicated in the pathophysiology of several diseases;
specifically, NOXs in endothelial cells (ECs) are involved in
various vascular diseases (9). ECs
express four isoforms of NOX: Superoxide-generating enzyme NOX1,
NOX2, hydrogen peroxide-generating enzyme NOX4 and NOX5. NOX4 has
been implicated in EC apoptosis during the development of
bleomycin-induced lung fibrosis. It was also recently reported that
a NOX inhibitor reduced RIPF through inducing airway
epithelial-cell senescence (10).
Radiation-induced vascular damage has an important
role in normal tissue injury. Furthermore, EC dysfunction is
thought to be associated with thromboresistance, the inflammatory
response and vascular fibrosis (11–13).
The present study focused on the effects of NOXs on the
fibroblastic changes in ECs during RIPF. A specific NOX isoform
that regulates radiation-induced fibroblastic changes in ECs was
identified and the therapeutic potential of NOX inhibition in RIPF
was demonstrated.
Materials and methods
Cell culture and treatment
Human pulmonary artery endothelial cells (HPAECs)
were obtained from PromoCell (Heidelberg, Germany). All cells were
used within nine passages. Endothelial Cell Growth Medium 2
(PromoCell) was used for HPAEC culture. VAS2870 was purchased from
EMD Millipore (Billerica, MA, USA). Lentiviral vectors containing
small hairpin RNAs (shRNAs) targeting NOX1, -2 or -4, along with a
control shRNA (cat. nos. sc-43938-V, sc-35503-V, sc-41586-V, and
sc-108080), were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Transfection was performed using
5×104 infectious units of the respective vector for
6×105 cells. Lentiviral particles were directly added
into cells in OPTI-MEM (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and cells were incubated for 5 h with gentle
shaking every 30 min. Following the addition of growth medium,
cells were incubated for 2~3 days before further treatments. Cells
were exposed to gamma rays derived from a [137Cs] source
using GammaCell 3000 (Atomic Energy of Canada, Mississauga, OT,
Canada) at a dose rate of 3.81 Gy/min.
Measurement of ROS levels
Following the indicated treatments, cells were
incubated for 30 min with 1 µm
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Invitrogen; Thermo Fisher Scientific, Inc.) or 2.5 µm
MitoSOX™ (Invitrogen) at 37°C, and washed twice with
phosphate-buffered saline (PBS). The samples were then re-suspended
in 1 ml PBS and analyzed using a BD FACScan flow cytometer (BD
Biosciences, Franklin Lakes, NJ, USA).
Immunofluorescence staining
Following the indicated treatments, cells were fixed
with 100% ice-cold acetone (Sigma-Aldrich, St. Louis, MO, USA) for
5 min, washed three times with PBS (pH 7.3) and incubated with
antibodies against α-SMA (1:1,000; mouse monoclonal anti-human; cat
no. A5228; Sigma-Aldrich) and CD31 (1:100; goat polyclonal anti
human; cat no. sc-1506; Santa Cruz), VE-cadherin (1:100; mouse
monoclonal anti-human; cat no. sc-9989; Santa Cruz), and FSP
(1:100; rabbit polyclonal anti-human, cat no. ab27957; Abcam,
Cambridge, UK) in PBS containing Tween 20 (PBST; Sigma-Aldrich) and
2% bovine serum albumin (Sigma-Aldrich) for 3 h. Subsequently,
samples were incubated with Alexa 488-conjugated donkey anti-mouse
(or rabbit) and Alexa 546-conjugated donkey anti-goat (or mouse)
antibodies (1:250; cat no. A21202, A21206, A11056, and A10036 ;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) for 1 h and
washed with PBS. Cell nuclei were labeled with DAPI (5 µM;
cat no. D9542; Sigma-Aldrich), and stained cells were imaged using
a Zeiss confocal microscope (Carl Zeiss AG, Oberkochen, Germany;
magnification, ×400).
Western blot analysis
For Western blot analysis, cells were lysed with
RIPA (radio immunoprecipitation assay) buffer [50 mM Tris-HCl (pH
7.5), 150 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), and
1% sodium deoxycholate] supplemented with 1 mM
Na3VO4, 1 mM dithiothreitol, 1 mM
phenylmethylsulfonyl fluoride, and protease inhibitor cocktail (EMD
Millipore). The supernatants were collected after centrifugation at
16,000 × g for 15 min at 4°C. Protein concentrations were measured
using Bio-Rad Protein Assay (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The samples were boiled for 5 min, and an equal amount of
protein was separated using SDS-polyacrylamide gel electrophoresis
and transferred onto BioTrace NT Nitrocellulose Transfer Membranes
(Pall Corporation, Pensacola, FL, USA). The membranes were
incubated overnight at 4°C with primary antibodies against CD31
(1:1,000; cat=o. sc-1506, goat polyclonal anti-human), vascular
endothelial (VE)-cadherin (1:1,000; cat no. sc-9989, mouse
monoclonal anti-human), TGF-β-receptor kinase I (ALK5) (1:1,000;
cat no. sc-398, rabbit polyclonal anti-human), vimentin (1:1,000;
cat no. sc-6260, mouse monoclonal anti-human), intercellular
adhesion molecule 1 (ICAM1; 1:1,000; cat no. sc-7891, mouse
anti-human) (all from Santa Cruz Biotechnology, Inc.), α-SMA
(1:5,000; cat no. A5228; mouse monoclonal anti-human) and β-actin
(1:5,000; cat no. A1978, mouse monoclonal anti-human). All
antibodies were sourced from Sigma-Aldrich. The membranes were then
incubated with HRP-conjugated donkey anti-goat, goat anti-mouse,
and goat anti-rabbit secondary antibodies (1:4000; cat nos.
sc-2020, sc-2005, and sc-2004) for 1 h at room temperature. HRP
activity was measured using Western Lightning Plus-ECL
(PerkinElmer, Inc., Waltham, MA, USA) and protein band intensity
was visualized on AGFA CP-BU Medical X-Ray Film (Agfa HealthCare
NV, Gevaert, Belgium) and quantified using Image J software 1.45
(National Institutes of Health, Bethesda, MD, USA)
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol
reagent (MRC, Cincinnati, OH, USA) and 1 µg of total RNA was
used to synthesize cDNA with an Omniscript RT kit (Qiagen, Hilden,
Germany) according to the manufacturer's protocol. Then, 2
µl of cDNA was amplified with Solgent Taq (Solgent, Daejeon,
Korea) kits in a total volume of 25 µl according to the
manufacturer's protocol. The primers for NOX1, NOX2, and NOX4 were
synthesized by Integrated DNA Technologies, Inc. (Coralville, IA,
USA). PCR products were detected in agarose gels containing 1
µg/ml ethidium bromide. The primer sequences used were as
follows. NOX1 forward, 5′-TGGAGTGGCTTGCACC-3′ and reverse,
5′-TGCTGCATGACCAACCTTTT-3′; NOX2 forward,
5′-TTTACACTGACATCCGCCCC-3′ and reverse, 5′-TGGGCCGTCCATACAAAGTC-3′;
NOX4 forward, 5′-CGGGCTTCCACTCAGTCTTT-3′ and reverse,
5′-TGATCCGAGGTAAGCCA-3′. PCR conditions were as follows: 95°C for 2
min, followed by 35 cycles, denaturation at 95°C for 30 sec,
annealing at 58°C for 30 sec, and extension at 72°C for 45 sec. PCR
products were detected in 2% agarose gels containing 1 µg/ml
ethidium bromide, and scanned by Gel Doc™ XR+ Imager System
(Bio-Rad Laboratories, Inc.). Band intensities of PCR products were
quantified using Image J software (version 1.45; National
Institutes of Health, Bethesda, MD, USA). The 2−ΔΔCq
method was used to calculate the band density (14).
Mice and irradiation
All procedures of the present study were approved by
the Institutional Animal Care and Use Committee of the Korea
Institute of Radiological and Medical Sciences (Seoul, Korea). Mice
(weight, 25–30 g) were purchased from Orient Bio (Seoul, South
Korea) and maintained in 12 h light 12 h dark cycle with access to
food and water ad libitum. They were maintained in an
atmosphere of 18–24°C, with 40–60% humidity. A total of 28 mice
were used (control group, n=4; 25 Gy group, n=5, 25 Gy + NOX
inhibitor group, n=5; repeated twice). Radiation was delivered
using an X-RAD 320 platform (Precision X-ray, North Branford, CT,
USA) as described previously (15). Whole lungs of 10-week-old male
C57BL/6 mice were irradiated. An NOX1 VAS2870 inhibitor (EMD
Millipore) was dissolved in dimethyl sulfoxide (Sigma-Aldrich),
further diluted in distilled water and administered
intraperitoneally (25 mg/kg) 1 h prior to thoracic irradiation with
25 Gy; this was repeated twice at 1-day intervals.
Tissue histology and immunohistochemical
staining
Mice were sacrificed by anesthesia with
CO2. The lung tissue was harvested and fixed in 10%
(v/v) neutral buffered formalin (Sigma-Aldrich) prior to
preparation of paraffin (Sigma-Aldrich)-embedded samples, which
were cut into 3–4 µm sections. To detect collagen, sections
were de-paraffinized in xylene (Duksan Pure Chemicals Co., Ltd.,
Ansan, South Korea) and an ethanol series (95, 90, 70 and 50%;
Duksan Pure Chemicals Co., Ltd.), followed by washing with
phosphate-buffered saline (PBS; Welgene, Inc., Gyeongsan-si, South
Korea). De-paraffinized slides were boiled in 0.1 M citrate buffer
(pH 6.0; Target retrival solution; Dako, Glostrup, Germany) for 20
min and incubated in 3% hydrogen peroxide (Sigma-Aldrich) for 20
min. Slides were then stained using a Masson's Trichrome Stain kit
(Sigma-Aldrich) as described previously (16). Images were obtained using a Zeiss
microscope (Carl Zeiss AG).
For immunofluorescence assays, de-paraffinized
slides were boiled in 0.1 M citrate buffer (pH 6.0) for 20 min and
incubated with 3% hydrogen peroxide for 20 min. Slides were
co-immunostained overnight at 4°C with a goat polyclonal anti-human
CD31 antibody (cat no. sc-1506; 1:100 dilution; Santa Cruz
Biotechnology, Inc.) and rabbit polyclonal anti-human alpha smooth
muscle actin (α-SMA) mAb (cat no. ab5694; 1:100 dilution; Abcam),
followed by incubation for 1 h at room temperature with donkey
anti-goat Alexa 488- or donkey anti-rabbit Alexa 546-conjugated
secondary antibodies (1:250; cat no. A11055 and A10040; Thermo
Fisher Scientific, Inc.). Cell nuclei were labeled with DAPI (5
µM; cat no. D9542; Sigma-Aldrich). Images were captured
using a Zeiss confocal microscope. Image J software (version 1.45)
was used for quantitative evaluation of the staining.
Statistical analysis
Values are expressed as the mean ± standard
deviation of at least three independent experiments. Student's
t-tests and analysis of variance were used to explore the
statistical significance of differences between experimental
groups. Statistical analyses were performed using GraphPad Prism
software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
NOX inhibition reduces the generation of
ROS in HPAECs
It has been suggested that NOXs have important roles
in various pathological processes, including hypertension,
cardiovascular disease and stroke (9,17).
The present study therefore assessed whether ROS production during
radiation-induced lung damage was accountable for radiation-induced
late normal tissue injuries including atherosclerosis and fibrosis
in RIPF. Fluorescence-activated cell sorting using a DCF dye
indicated that NOX inhibition reduced radiation-induced ROS in
HPAECs (Fig. 1A). At 72 h after
irradiation with 5 Gy, the ROS content was elevated >2.0 fold,
which was, however, significantly attenuated in the group treated
with the NOX inhibitor VAS28701 (1 µm).
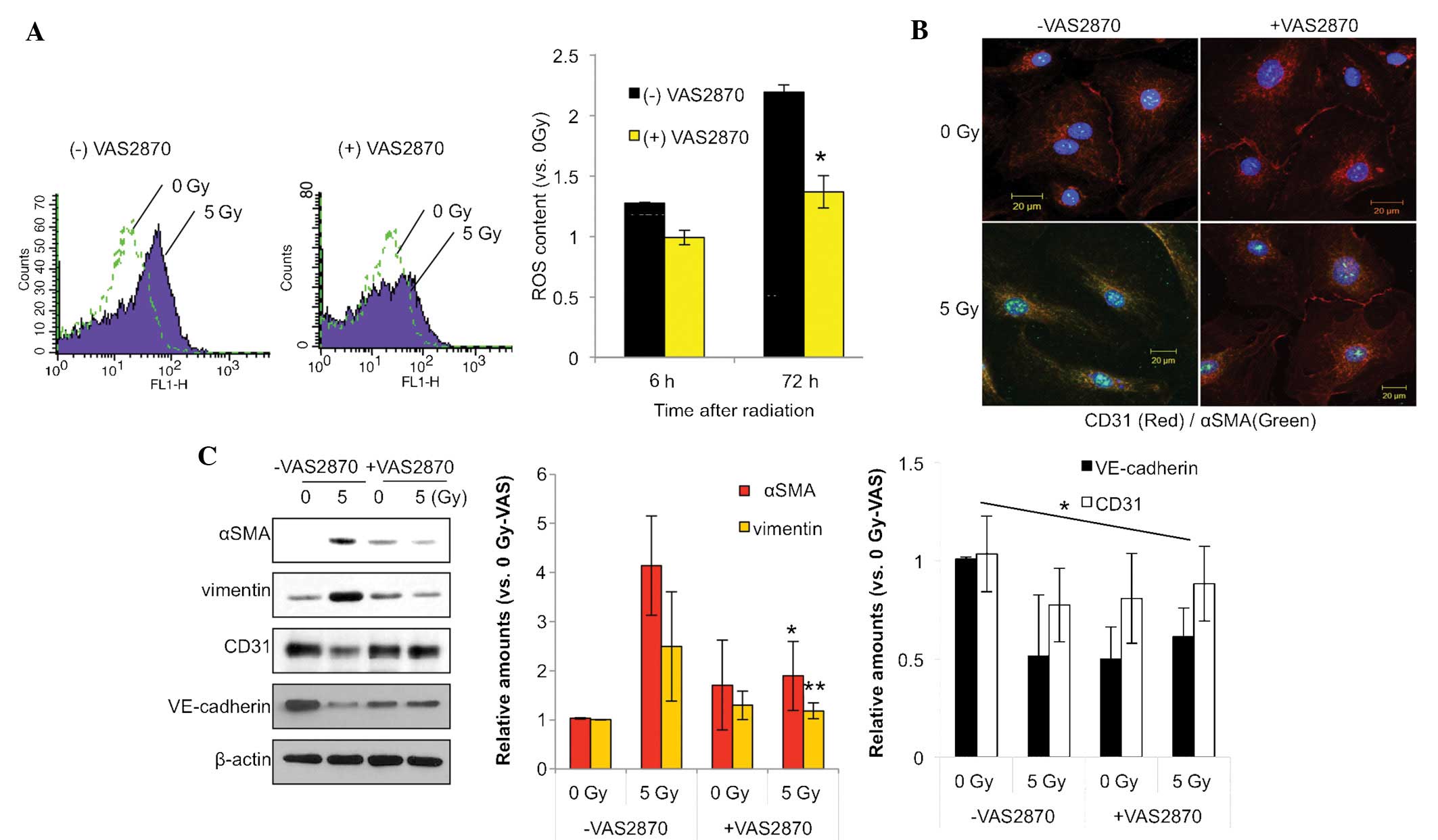 | Figure 1VAS2870 inhibits radiation-induced
fibroblastic changes in ECs. (A) HPAECs were irradiated with 5 Gy
and incubated for 72 h. VAS2870 (1 µm) was added to cells 1
h prior to irradiation. To measure ROS, cells were incubated for 30
min with 1 µm 2′,7′-dichlorodihydrofluorescein diacetate and
analyzed by flow cytometry (*P<0.05 vs. no VAS2870).
(B) HPAECs were irradiated with 5 Gy and incubated for 72 h.
VAS2870 (1 µm) was added to cells 1 h prior to irradiation
and analysis by immunofluorescence with Alexa 488-conjugated
anti-α-SMA and Alexa 594-conjugated anti-CD31 antibodies (green and
red, respectively). Nuclei were counterstained with
4′,6-diamidino-2-phenylindole (blue) (scale bars, 20 µm).
(C) Samples were subjected to western blot analysis of α-SMA,
vimentin, CD31 and VE-cadherin. β-actin served as the loading
control. Protein expression was quantified by densitometric
analysis. Values are expressed as the mean ± standard deviation
(n=3). *P<0.05 and **P<0.01 vs.
VAS2870-untreated. HPAEC, human pulmonary artery endothelial cell;
SMA, smooth-muscle actin; VAS, nicotinamide adenine dinucleotide
phosphate oxidase inhibitor VAS2870; ROS, reactive oxygen species;
VE, vascular endothelial. |
NOX inhibition reduces radiation-induced
fibrotic changes in HPAECs
As shown in Fig.
1B, immunofluorescence analysis revealed that radiation
increased the expression of α-SMA, a fibroblastic cell marker,
while decreasing the expression of EC marker CD31. Of note, these
effects were inhibited by treatment with NOX inhibitor VAS2870. The
endothelial-specific adhesion molecule CD31 was localized on the
membranes of un-irradiated HPAECs and disappeared as the cells
underwent fibroblastic changes, such as the
endothelial-to-mesenchymal transition, in response to irradiation
(Fig. 1B).
Consistent with this finding, western blot analysis
showed increased expression of the fibrotic markers α-SMA and
vimentin, and decreased expression of the EC markers CD31 and
VE-cadherin 3 days after 5-Gy radiation in HPAECs, which was
significantly inhibited by VAS2870 treatment (Fig. 1C). These results indicated that NOX
inhibition reduces ROS production and thus radiation-induced
fibroblastic changes in HPAECs.
shRNA-mediated knockdown of NOX1, -2 and
-4 decreases radiation-induced ROS in HPAECs
To determine which NOX isoform regulates the
radiation-induced ROS generation in ECs, their expression was
determined by RT-qPCR. Irradiation increased the expression levels
of NOX1, -2 and -4 on day 3 after 5-Gy radiation and to a lesser
extent on days 5 and 7 (Fig. 2A).
For further mechanistic study, HPAECs were transfected with NOX1,
-2 or -4 shRNA. The decreased expression of the respective NOXs
demonstrated the transfection efficiency of shRNA against NOX1, -2
or -4 (Fig. 2B). NOX1 shRNA
specifically decreased radiation-induced NOX1 expression, while
NOX2 and -4 shRNAs inhibited the expression of NOX2 as well as
NOX4, indicating that NOX2 and -4 are cross-regulated. The effects
of NOX knockdown on irradiation-induced ROS levels were then
assessed. The level of intracellular ROS and mitochondrial
superoxide (detected by H2DFHDA and mitSOX dye, respectively) in
irradiated HPAECs were reduced by pre-treatment with NOX1, -2 or 4
shRNA to a similar extent (Fig. 2C and
D). These results showed that knockdown of NOX1, -2 and -4
decreased radiation-induced ROS generation in HPAECs; furthermore,
NOX1-targeted shRNA was indicated to be a more specific regulator
than NOX2 or NOX4 shRNA.
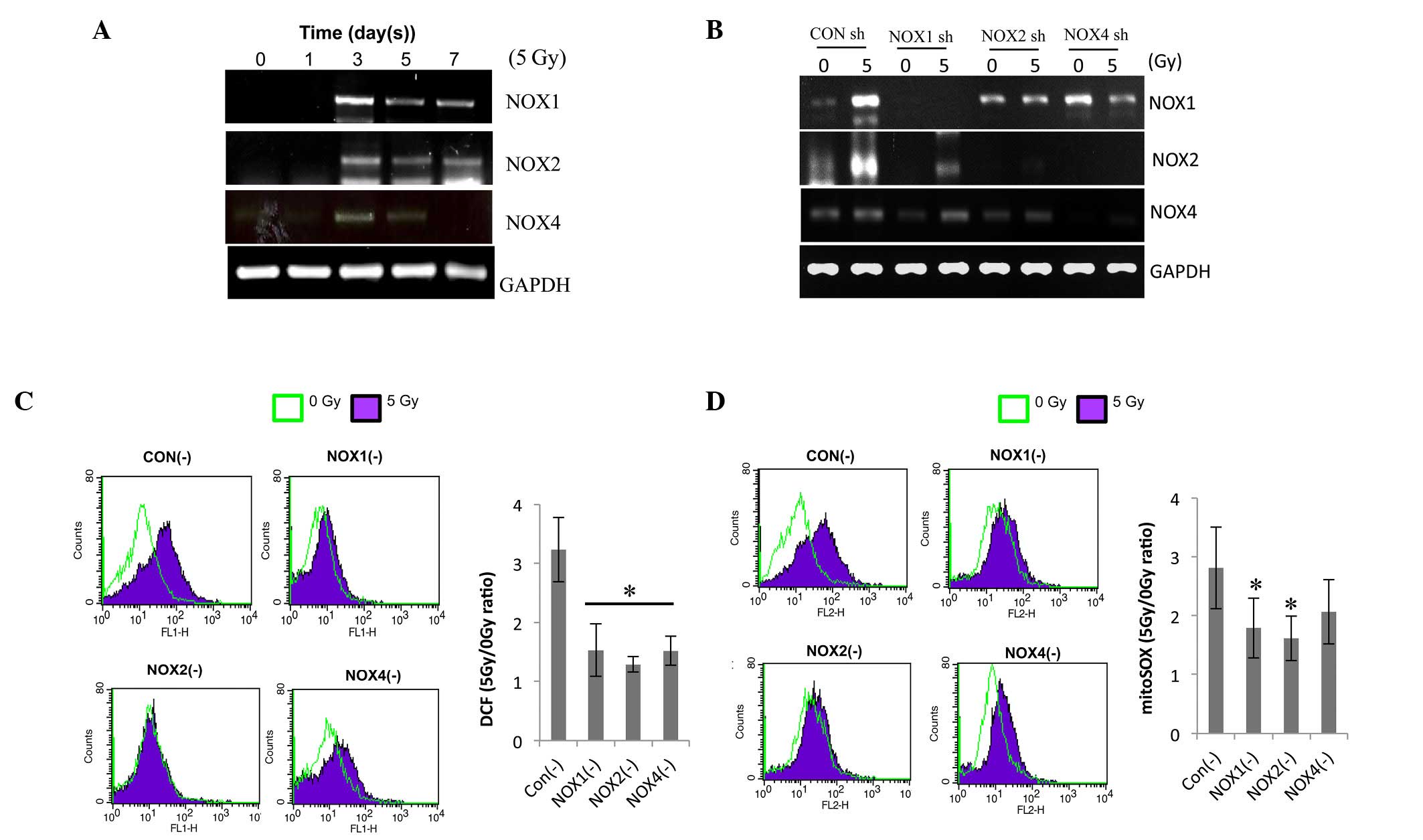 | Figure 2NOX1, 2 and 4 shRNAs decrease
radiation-induced ROS in HPAECs. (A) Following irradiation, NOX1, 2
and 4 expression was evaluated by RT-qPCR. HPAECs were cultured for
the indicated number of days after receiving 5 Gy irradiation. (B)
Each lentiviral vector contained a NOX1-, 2- or 4-targeted shRNA,
which was then transfected into cultured HPAECs. A lentiviral
vector containing a scrambled sequence served as the control. To
confirm lentiviral-mediated gene knockdown, NOX1, 2 and 4
expression was analyzed by RT-qPCR. (C) Assessment of ROS and (D)
determination of mitochondrial superoxide. Infected cells were
irradiated with 5 Gy, followed by incubation with 1 µm
H2DCFDA or 2.5 µm mitoSOX™, respectively, for 30
min and flow cytometric analysis. Values are expressed as the mean
± standard deviation. *P<0.05 (n=6) in C;
*P=0.05 (n=3) in D vs. control shRNA. HPAEC, human
pulmonary artery endothelial cell; NOX, nicotinamide adenine
dinucleotide phosphate oxidase; ROS, reactive oxygen species;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; shRNA, small
hairpin RNA; CON, control; RT-qPCR, reverse-transcription
quantitative polymerase chain reaction; H2DCFDA,
2′,7′-dichlorodihydrofluorescein diacetate. |
NOX1 shRNA decreases radiation-induced
fibrotic changes in HPAECs
Next, the present study aimed to identify the NOX
isoform that is accountable for radiation-induced fibrotic changes
in HPAECs. In HPAECs transfected with control shRNA, fibrotic
changes were observed, including increased expression levels of
α-SMA and vimentin as well as decreased CD31 expression (Fig. 3A). NOX1 shRNA more significantly
inhibited radiation-induced α-SMA compared with NOX2 or -4 shRNA
(Fig. 3A). In addition,
immunofluorescence analysis showed that the irradiation-induced
increase in fibroblast-specific protein 1 (FSP1), a fibroblastic
cell marker, and the decrease in VE-cadherin were inhibited by NOX1
knockdown (Fig. 3B).
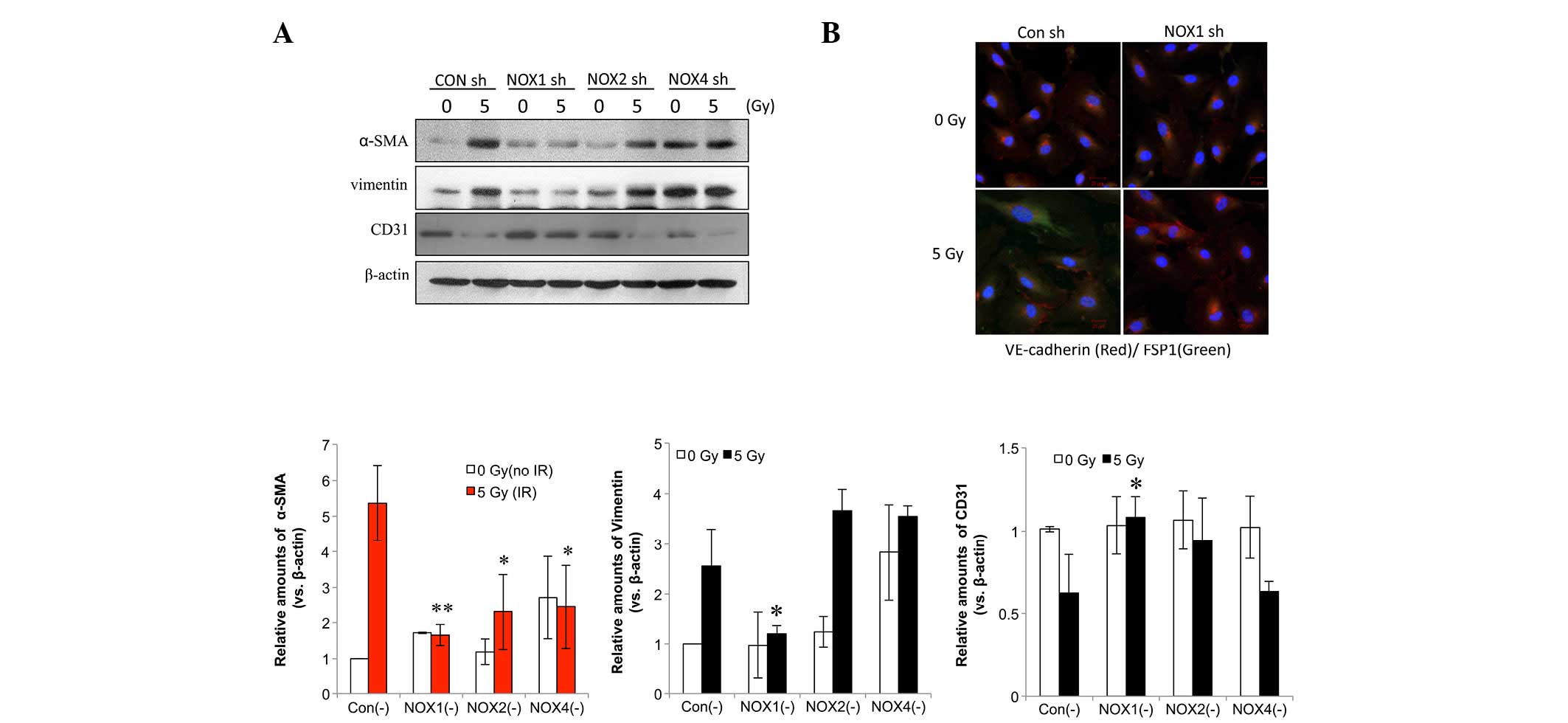 | Figure 3NOX1 shRNA decreases radiation-induced
fibrotic changes in HPAECs. (A) Cells transfected with NOX1-, 2- or
4-targeted shRNA were irradiated and incubated for 72 h, followed
by western blot analysis of α-SMA, vimentin and CD31. Protein
levels were quantified by densitometric analysis of the blots.
Values are expressed as the mean ± standard deviation (n=3).
**P<0.005; *P<0.05, α-SMA vs. Con (-). (B) HPAECs transfected
with NOX1 shRNA were irradiated with 5 Gy and incubated for 72 h.
Alexa 488-conjugated anti-FSP1 and Alexa 594-conjugated
anti-VE-cadherin antibodies (green and red, respectively) were used
to stain cells and nuclei were counterstained with
4′,6-diamidino-2-phenylindole (blue). HPAEC, human pulmonary artery
endothelial cell; SMA, smooth-muscle actin; VE, vascular
endothelial; NOX, nicotinamide adenine dinucleotide phosphate
oxidase; CON, control; shRNA, small hairpin RNA; IR, irradiation;
FSP fibroblast-specific protein. |
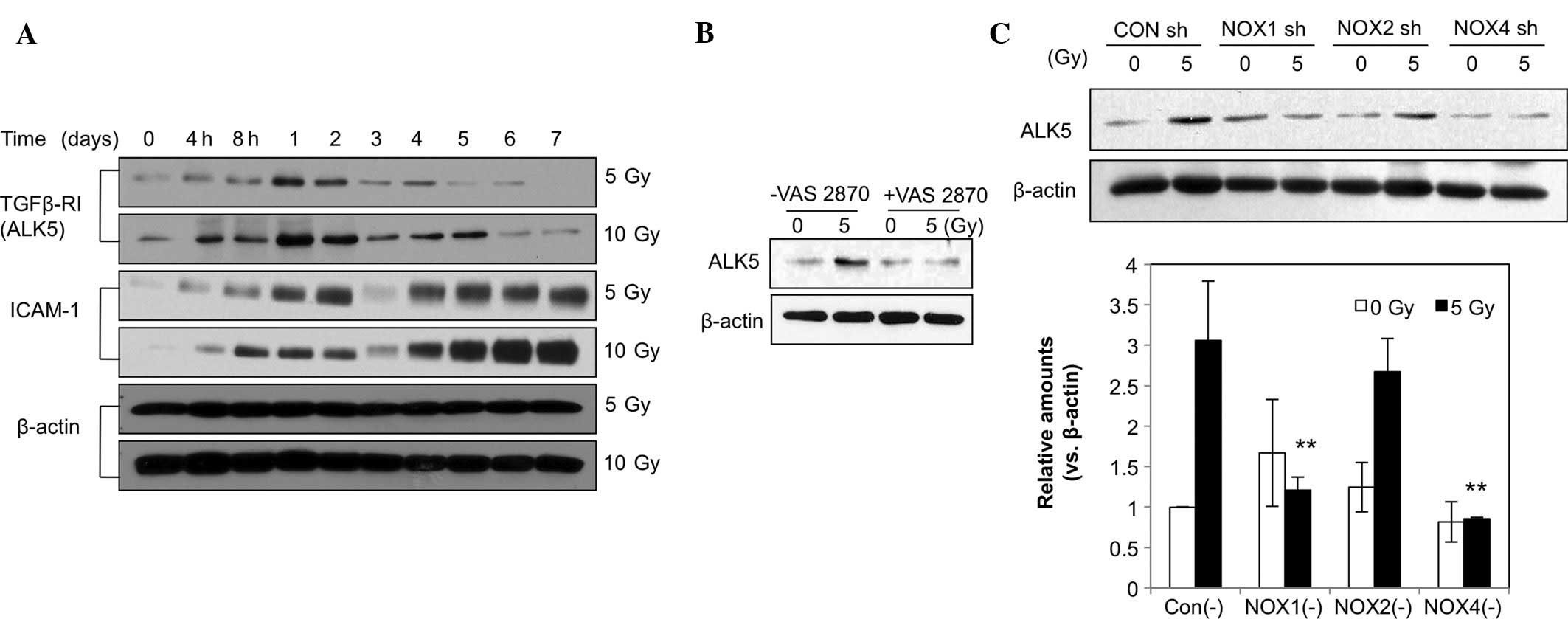
ALK5 expression was markedly increased at 4 h
post-irradiation and peaked after 1 day, followed by a gradual
decline until day 6, while the 10-Gy dose had a greater effect than
the 5-Gy dose (Fig. 4A). ICAM-1
levels were increased from 4 h on day 2 through day. 4 until day 7,
corresponding to the transient and late responses. ALK5-associated
signaling has been reported to mediate changes in the EC phenotype
(18). The elevated ALK5 levels
may also have been associated with the increased levels of the
fibroblastic markers α-SMA and vimentin. Furthermore, as shown in
Fig. 4A, radiation (5 or 10 Gy)
induced an increase in the levels of ICAM-1, indicating EC
activation and a phenotypic change. Increased ICAM-1 levels
appeared as a dual regulation from 4 h to day 2 and from days 4–7.
To assess the role of NOX in the radiation-induced increases in
ALK5, HPAECs were pre-treated with VAS2870. The results showed that
the increases in ALK5 levels caused by irradiation were markedly
attenuated by VAS2870 (Fig. 4B).
In addition, HPAECs were pre-treated with NOX1, -2 or -4 shRNA
prior to irradiation. As shown in Fig.
4C, pre-treatment with NOX1 shRNA significantly inhibited
irradiation-induced increases in ALK5 expression (Fig. 4C). These results suggested that
NOX1 is a molecule involved in the regulation of radiation-induced
fibrotic changes in ECs via ALK5 signaling. ROS generated by NOX1
may contribute to fibrotic changes in irradiated ECs.
NOX1 inhibition reduces radiation-induced
collagen deposition in the lung
An in vivo experiment was employed to
demonstrate that an NOX1-specific inhibitor reduced
radiation-induced collagen deposition during the development of
RIPF (Fig. 5A). C57BL/6 mice
received 25-Gy irradiation to the thoracic region with or without
pre-treatment with NOX1-specific inhibitor. In
inhibitor-pre-treated animals, the NOX1 inhibitor was further
administered twice at 2-day intervals by intraperitoneal injection.
Four weeks after irradiation, collagen deposition in the irradiated
lung tissues was analyzed by trichrome staining. As shown in
Fig. 5B, collagen deposition was
increased in the irradiated lung tissues, while pre-treatment with
NOX1 inhibitor significantly decreased collagen deposition.
To further examine fibroblastic changes in the ECs
of the irradiated lung tissues, immunofluorescence assays were
performed. Similar to the in vitro data, α-SMA was
upregulated and co-localized with CD31 in the ECs of irradiated
lung tissues, indicating fibrotic changes (Fig. 5C). In addition, the NOX1 inhibitor
abrogated the increases in α-SMA expression in these ECs (Fig. 5C). It was therefore suggested that
the observed fibrotic changes in the irradiated lung tissues may
have contributed to increased collagen deposition. Furthermore,
these results indicated that endothelial NOX1 inhibition can
specifically diminish RIPF via attenuation of fibroblastic changes
in irradiated ECs.
Discussion
RIPF generally develops ~6–24 months following
tissue damage and stabilizes after two years (1). This chronic complication is thought
to be caused by chronic ROS accumulation (6,19).
Specifically, NOXs generate superoxide, a toxic type of ROS. Since
NOXs are constitutively present in most cell types, NOX inhibitors
are associated with toxic effects (9,17). A
recent study showed that NOX blocked the radiation-mediated
upregulation of intracellular ROS in microvascular ECs of the rat
brain, suggesting that NOX may be an important regulator of
radiation-induced brain injury in patients with brain metastasis
(20). Although NOX is an
efficient target for regulating ROS in various diseases, including
radiation-induced tissue damage, its clinical use is limited by the
unpredictable side effects of non-selective NOX inhibition
(21). Thus, the present study
aimed to identify the specific NOX isoform that regulates RIPF. The
role of NOX in RIPF has remained to be fully elucidated. Recently,
Jarman et al (22) reported
that an NOX4 inhibitor reduced idiopathic pulmonary fibrosis
through a TGF-β-associated signaling mechanism.
Radiation-induced late normal tissue injuries
including atherosclerosis and fibrosis are, in part, due to
vascular compromise. A recent study by our group reported
fibroblastic changes in vascular ECs in radiation-induced
atherosclerosis (23). In
addition, vascular damage and subsequent inflammation can occur
during RIPF development (13).
During RIPF development, radiation can disrupt the integrity of the
pulmonary epithelium and endothelium, leading to edema and
leukocyte recruitment, and resulting in alterations of the
microenvironment (24).
Several studies have reported radiation-induced
fibroblastic phenotypic changes in alveolar epithelial cells
(10,17). Furthermore, mesenchymal transition
or senescence of alveolar epithelial cells are linked with the
development of RIPF (10,25). However, the underlying molecular
mechanisms of pathological changes in vascular ECs resulting in
chronic fibrosis in radiation-induced lung injury have remained
elusive. Adamson and Bowden (26)
suggested that acute endothelial injury may be rapidly repaired
with little stimulation of the fibroblasts, while more severe or
prolonged injury with delayed regeneration disrupting the
endothelial-mesenchymal balance. The endothelial-to-mesenchymal
transition is thought to be mainly associated with diseases
including cardiac fibrosis, kidney fibrosis, bleomycin-induced
fibrosis and cancer (19,22). However, the mechanism underlying
the fibroblastic changes that occur in ECs during RIPF have
remained elusive.
The present study identified fibroblastic changes in
vascular HPAECs subjected to irradiation, which promoted collagen
deposition. Specifically, NOX1 shRNA efficiently inhibited fibrotic
changes in ECs compared with NOX2 and -4 shRNA in irradiated
HPAECs. NOX1 has been reported to be expressed in vascular smooth
muscle cells and ECs (9).
Furthermore, NOX1 is more closely associated with EC dysfunction in
ROS-induced acute injury than other NOX isoforms (9,17,27).
Several studies have reported that basal blood pressure, systemic
hypertension and the early stage of atherosclerosis depend on NOX1
(9,17,27).
In the present study, as expected, a small NOX1-specific inhibitor
significantly inhibited RIPF in vivo. Therefore, it is
hypothesized that NOX1 is a specific target in the regulation of
radiation-induced EC damage and subsequent development of RIPF. In
addition, it is indicated that fibroblastic changes in irradiated
ECs may be accountable for RIPF in the initial stage of
radiation-induced lung damage.
Acknowledgments
The present study was supported by the Nuclear
Research and Development Program (grant nos. NRF-2012M2A2A7012483,
NRF-2011-0031697 and NRF-2013M2A2A7043580) and a KIRAMS Research
Project (grant no. 50520-2013) funded by the Nuclear Research and
Development Program.
References
|
1
|
Mehta V: Radiation pneumonitis and
pulmonary fibrosis in non-small-cell lung cancer: Pulmonary
function, prediction and prevention. Int J Radiat Oncol Biol Phys.
63:5–24. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleckenstein K, Gauter-Fleckenstein B,
Jackson IL, Rabbani Z, Anscher M and Vujaskovic Z: Using biological
markers to predict risk of radiation injury. Semin Radiat Oncol.
17:89–98. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K,
Matsui H, Hara K, Tanaka T, Iso T, Suga T and Kurabayashi M:
Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production
in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung
Cell Mol Physiol. 300:L740–L752. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rodemann HP and Bamberg M: Cellular basis
of radiation-induced fibrosis. Radiother Oncol. 35:83–90. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Molteni A, Moulder JE, Cohen EF, Ward WF,
Fish BL, Taylor JM, Wolfe LF, Brizio-Molteni L and Veno P: Control
of radiation-induced pneumopathy and lung fibrosis by
angiotensin-converting enzyme inhibitors and an angiotensin II type
1 receptor blocker. Int J Radiat Biol. 76:523–532. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guerrero T, Martinez J, McCurdy MR, Wolski
M and McAleer MF: Elevation in exhaled nitric oxide predicts for
radiation pneumonitis. Int J Radiat Oncol Biol Phys. 82:981–988.
2012. View Article : Google Scholar
|
|
7
|
Zhang Y, Zhang X, Rabbani ZN, Jackson IL
and Vujaskovic Z: Oxidative stress mediates radiation lung injury
by inducing apoptosis. Int J Radiat Oncol Biol Phys. 83:740–748.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fleckenstein K, Zgonjanin L, Chen L,
Rabbani Z, Jackson IL, Thrasher B, Kirkpatrick J, Foster WM and
Vujaskovic Z: Temporal onset of hypoxia and oxidative stress after
pulmonary irradiation. Int J Radiat Oncol Biol Phys. 68:196–204.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Drummond GR and Sobey CG: Endothelial
NADPH oxidases: Which NOX to target in vascular disease? Trends
Endocrinol Metab. 25:452–463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Citrin DE, Shankavaram U, Horton JA,
Shield W III, Zhao S, Asano H, White A, Sowers A, Thetford A and
Chung EJ: Role of type II pneumocyte senescence in
radiation-induced lung fibrosis. J Natl Cancer Inst. 105:1474–1484.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brush J, Lipnick SL, Phillips T, Sitko J,
McDonald JT and McBride WH: Molecular mechanisms of late normal
tissue injury. Semin Radiat Oncol. 17:121–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yarnold J and Brotons MC: Pathogenetic
mechanisms in radiation fibrosis. Radiother Oncol. 97:149–161.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weintraub NL, Jones WK and Manka D:
Understanding radiation-induced vascular disease. J Am Coll
Cardiol. 55:1237–1239. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hong ZY, Lee HJ, Choi WH, Lee YJ, Eun SH,
Lee JI, Park K, Lee JM and Cho J: A preclinical rodent model of
acute radiation-induced lung injury after ablative focal
irradiation reflecting clinical stereotactic body radiotherapy.
Radiat Res. 182:83–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YJ, Koch M, Karl D, Torres-Collado AX,
Fernando NT, Rothrock C, Kuruppu D, Ryeom S, Iruela-Arispe ML and
Yoon SS: Variable inhibition of thrombospondin 1 against liver and
lung metastases through differential activation of
metalloproteinase ADAMTS1. Cancer Res. 70:948–956. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sanders KA and Hoidal JR: The NOX on
pulmonary hypertension. Circ Res. 101:224–226. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ten Dijke P, Egorova AD, Goumans MJ,
Poelmann RE and Hierck BP: TGF-β signaling in
endothelial-to-mesenchymal transition: The role of shear stress and
primary cilia. Sci Signal. 5:pt22012. View Article : Google Scholar
|
|
19
|
Mancini ML and Sonis ST: Mechanisms of
cellular fibrosis associated with cancer regimen-related
toxicities. Front Pharmacol. 5:512014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Collins-Underwood JR, Zhao W, Sharpe JG
and Robbins ME: NADPH oxidase mediates radiation-induced oxidative
stress in rat brain microvascular endothelial cells. Free Radic
Biol Med. 45:929–938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aldieri E, Riganti C, Polimeni M, Gazzano
E, Lussiana C, Campia I and Ghigo D: Classical inhibitors of NOX
NAD (P) H oxidases are not specific. Cur Drug Metab. 9:686–696.
2008. View Article : Google Scholar
|
|
22
|
Jarman ER, Khambata VS, Cope C, Jones P,
Roger J, Ye LY, Duggan N, Head D, Pearce A, Press NJ, et al: An
inhibitor of NADPH oxidase-4 attenuates established pulmonary
fibrosis in a rodent disease model. Am J Respir Cell Mol Biol.
50:158–169. 2014.
|
|
23
|
Kim M, Choi SH, Jin YB, Lee HJ, Ji YH, Kim
J, Lee YS and Lee YJ: The effect of oxidized low-density
lipoprotein (ox-LDL) on radiation-induced
endothelial-to-mesenchymal transition. Int J Radiat Biol.
89:356–363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rubin P, Johnston CJ, Williams JP,
McDonald S and Finkelstein JN: A perpetual cascade of cytokines
postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol
Biol Phys. 33:99–109. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Almeida C, Nagarajan D, Tian J, Leal SW,
Wheeler K, Munley M, Blackstock W and Zhao W: The role of alveolar
epithelium in radiation-induced lung injury. PloS One.
8:e536282013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adamson IY and Bowden DH: Endothelial
injury and repair in radiation-induced pulmonary fibrosis. Am J
Pathol. 112:224–230. 1983.PubMed/NCBI
|
|
27
|
Ray R and Shah AM: NADPH oxidase and
endothelial cell function. Clin Sci (Lond). 109:217–226. 2005.
View Article : Google Scholar
|