Introduction
Reactive oxygen species (ROS) are biologically
significant due to their role in cellular redox signaling (1). It has been reported that ROS may
induce cellular damage and increase physiological dysfunction
(2). In locations where ROS
accumulate, oxidative damage occurs, which has been linked to a
diverse range of neurodegenerative disorders, including Alzheimer's
disease, Parkinson's disease, amyotrophic lateral sclerosis, prion
diseases, hereditary ataxia, dentatorubral-pallidoluysian atrophy
and Wilson's disease, as well as cancer and skin aging (3,4).
Glutamate is the fundamental excitatory neurotransmitter, which is
activated via N-methyl-D-aspartate receptors. Several important
physiological functions are co-regulated by glutamate, and
excessive concentrations of glutamate lead to the pathological
effects caused by ROS. In addition, glutamate-associated
neurotoxicity is implicated in numerous neuronal disorders
(5). Hydrogen peroxide
(H2O2) is the product of a non-radical
two-electron reduction of oxygen, which has previously been
implicated in redox signaling and oxidative stress (6). Heme consists of Fe2+ and
protoporphyrin IX, and is a prosthetic group that is present in
hemoglobin and myoglobin (7). Free
heme can lead to oxidative damage; therefore, in order to degrade
heme, cells produce the rate-limiting enzyme heme oxygenase (HO).
HO-1, which is an inducible form of HO, degrades heme into three
byproducts: Biliverdin, carbon monoxide (CO) and Fe2+.
CO is generally considered to be toxic; however, recent studies
have suggested that it exerts antiproliferative, anti-inflammatory
and anti-apoptotic effects (8–10).
Biliverdin is converted into bilirubin by biliverdin reductase, and
numerous reports have suggested that bilirubin exerts antioxidant
effects (11,12). Fe2+ is immediately
converted to ferritin, which has a protective effect against heme
synthesis (13). Nuclear factor
erythroid-derived 2-related factor-2 (Nrf2) has been reported to
act as a positive regulator of detoxification enzyme gene
expression, and recent studies have demonstrated that Nrf2
translocation regulates the expression of hundreds of
cytoprotective genes, which counteract endogenously- or
exogenously-generated oxidative stress (14). Nrf2 is a major upstream donor that
induces HO expression (15).
Mitogen-activated protein kinases (MAPKs) are associated with the
majority of signal transduction pathways, including those involved
in cell differentiation, cell proliferation, cell survival and cell
transformation (16). The MAPK
family comprises extracellular signal-regulated kinase (ERK), c-Jun
NH2-terminal kinase (JNK) and p38 MAPK. Previous studies have
demonstrated that MAPK is activated by oxidative stress or other
stimuli, and that phosphorylation of MAPK regulates the expression
of diverse genes and proteins, including HO-1 (17,18).
KCHO-1 is a novel mixture comprised of 30% ethanol
(EtOH) extracts obtained from nine natural products: Curcuma longa,
Salvia miltiorrhiza, Gastrodia elata, Chaenomeles sinensis,
Polygala tenuifolia, Paeonia japonica, Glycyrrhiza uralensis,
Atractylodes japonica and processed Aconitum carmichaeli. These
natural products are well known as traditional medicinal herbs,
which are used as alternative therapies in Korea and China, and
recent studies have reported the beneficial effects of these herbs
(19–25). In our previous study, it was
suggested that KCHO-1 exerted anti-inflammatory effects in BV2
microglia (26). Using an in vitro
oxidative stress model, the present study aimed to explore the
direct neuroprotective effects of KCHO-1, and to determine the
possible underlying mechanisms.
Materials and methods
Reagents
Dulbecco's modified Eagle's medium (DMEM) and other
tissue culture reagents were purchased from Gibco (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The HO activity inhibitor tin
protoporphyrin IX (SnPP IX) was obtained from Porphyrin Products
(Frontier Scientific, Logan, UT, USA). Primary antibodies,
including rabbit polyclonal anti-HO-1 (1:1,000 dilution; cat. no.
sc-10789), rabbit polyclonal anti-Nrf2 (1:1,000 dilution; cat. no.
sc-722), goat polyclonal anti-lamin B (1:1,000 dilution; cat. no.
sc-6216) and goat polyclonal anti-actin (1:1,000 dilution; cat. no.
sc-1616) were purchased from Santa Cruz Biotechnology, Inc.
(Heidelberg, Germany). Rabbit polyclonal anti-phosphorylated-ERK
(1:1,000 dilution; cat. no. 9101) and rabbit polyclonal anti-ERK
(1:1,000 dilution; cat. no. 9102) antibodies were obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Secondary
horseradish peroxidase (HRP)-conjugated polyclonal goat anti-rabbit
IgG (1:1,000 dilution; cat. no. sc-2004) and HRP-conjugated normal
goat IgG (1:1,000 dilution; cat. no. sc-2741) were purchased from
Santa Cruz Biotechnology, Inc. The HO-1 inducer cobalt
protoporphyrin IX (CoPP) and all other chemicals used were obtained
from Sigma-Aldrich (St. Louis, MO, USA).
Extract preparation
C. longa, C. sinensis, P.
tenuifolia, P. japonica, G. uralensis and A.
japonica were purchased from Won Kwang Herb Co., Ltd. (Jinan,
South Korea) in August 2013. S. miltiorrhiza and G.
elata were purchased from Dongkyung Pharm. Co., Ltd. (Boeun,
South Korea). Processed A. carmichaeli was purchased from
Hanpoong Pharm & Foods Co., Ltd. (Jeonju, South Korea). All
voucher specimens were deposited at Hanpoong Pharm & Foods Co.,
Ltd. [C. longa (HP2013-10-01), S. miltiorrhiza
(HP2013-10-02), G. elata (HP2013-10-03), C. sinensis
(HP2013-10-04), P. tenuifolia (HP2013-10-05), P.
japonica (HP2013-10-06), G. uralensis (HP2013-10-07),
A. japonica (HP2013-10-08), and processed A.
carmichaeli (HP2013-10-09)]. To prepare the extract, C.
longa (4 kg), S. miltiorrhiza (4 kg), G. elata (4
kg), C. sinensis (2 kg), P. tenuifolia (2 kg), P.
japonica (2 kg), G. uralensis (2 kg), A. japonica
(2 kg) and processed A. carmichaeli (1 kg) were mixed,
pulverized and extracted in 30% EtOH for 3 h at 84–90°C.
Subsequently, the mixture was concentrated using a rotary
evaporator and lyophilized.
High-performance liquid chromatography
(HPLC) analysis
The sample was analyzed by reversed-phase HPLC using
a Sykam HPLC (Sykam GmbH, Eresing, Germany), equipped with S7131
Reagent Organizer, S2100 Solvent Delivery system, S7511 Vacuum
Degaser, S5200 Sample Injection and S3210 UV/Vis Detector.
HPLC-grade acetonitrile was purchased from Burdick &
Jackson® (Honeywell; Muskegon, MI, USA). Data processing
was carried out using ChromStar DAD (GPC) software (Sykam GmbH). An
Inertsil-ODS3 column (150×4.6 mm; particle size, 5 μm; GL
Sciences Inc., Torrance, CA, USA) was used in the stationary phase.
The mobile phase consisted of eluent A (0.1% formic acid in water
with 10% acetonitrile) and eluent B (acetonitrile). The starting
eluent was 100% A. The proportion of eluent B was increased
linearly to 36% from 0 to 60 min, increased to 60% from 60 to 90
min, and increased to 100% from 90 to 110 min. The detector
wavelength was set over a range of 190–700 nm and recorded at 254
nm. The flow rate was 1.0 ml/min, and the injection volume was 20
μl. Identification was based on comparison of retention time
and ultraviolet (UV) spectra with commercial standards. For each
compound, peak areas were determined as the wavelength providing
maximal UV absorbance.
Cell culture and viability assay
The HT22 mouse hippocampal cells were provided by
Dr. Inhee-Mook (Seoul National University, Seoul, South Korea). The
cells were maintained in DMEM supplemented with 10%
heat-inactivated fetal bovine serum, penicillin G (100 units/ml),
streptomycin (100 mg/ml) and L-glutamine (2 mM), and were incubated
at 37°C in a humidified atmosphere containing 5% CO2 and
95% air. For determination of cell viability, HT22 cells
(1×105 cells/well in 24-well plates) were incubated with
glutamate (0.5–20 mM; Sigma-Aldrich) and H2O2
(10–500 μM) for 12 h, or pre-treated with KCHO-1 (10–200
μg/ml; Sigma-Aldrich) for 12 h. SnPP IX (50 μM;
Sigma-Aldrich) was used as an inhibitor of HO, and trolox (50
μM) was used as a positive control, incubated with
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) at a final concentration of 0.5 mg/ml for 4 h.
Subsequently, the formazan that had formed was dissolved in acidic
2-propanol. Optical density (OD) was measured at 590 nm using a
microplate reader (model no. 680; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The OD of formazan in the control (untreated)
cells was considered to represent 100% viability.
ROS measurement
To measure ROS, HT22 cells (2.5×104
cells/well in 24-well plates) were treated with 5 mM glutamate (5
mM) in the presence or absence of KCHO-1 (10–200 μg/ml) or
SnPP IX (50 μM) for 8 h. After washing with
phosphate-buffered saline (PBS), the cells were stained with 10
μM 2′,7′-dichlorofluorescein diacetate in Hank's balanced
salt solution for 30 min in the dark. The cells were then washed
twice with PBS and extracted with 1% Triton X-100 in PBS for 10 min
at 37°C. Fluorescence was recorded at an excitation wavelength of
490 nm and an emission wavelength of 525 nm (Spectramax Gemini XS;
Molecular Devices, Sunnyvale, CA, USA). Cells were immediately
observed under a laser-scanning confocal microscope (TCS SP2; Leica
Microsystems, Wetzlar, Germany). Dichlorofluorescein fluorescence
was excited at 488 nm with an argon laser, and the resulting
emission was filtered with a 515-nm long pass filter.
Western blot analysis
HT22 cells were treated with KCHO-1 (10–200
μg), harvested and pelleted by centrifugation at 200 × g for
3 min. Subsequently, the cells were washed with PBS and lysed using
radioimmunoprecipitation assay lysis buffer [25 mmol/l Tris-HCl
buffer, pH 7.6; 150 mmol/l NaCl; 1% NP-40; 1% sodium deoxycholate;
0.1% sodium dodecyl sulfate (SDS)]. Protein concentration was
determined using Bradford Assay Reagent (Bio-Rad Laboratories,
Inc.). An equal amount of protein (30 μg) from each sample
was separated by 12% SDS-polyacrylamide gel electrophoresis and was
then electrophoretically transferred onto a Hybond-enhanced
chemiluminescence (ECL) nitrocellulose membrane (Bio-Rad
Laboratories, Inc.). The membrane was blocked with 5% skim milk and
incubated with primary antibodies at 4°C overnight, then incubated
with secondary antibodies at room temperature for 1 h. The bands
were then visualized using ECL (RPN2232; GE Healthcare Life
Sciences, Chalfont, UK) and quantified by densitometry using Image
J software (version 1.47; National Institutes of Health, Bethesda,
MA, USA). In the figures, representative blots from three
independent experiments are presented, and the data are presented
as the mean ± standard deviation of three independent experiments.
Nuclear and cytoplasmic cell extracts were prepared using NE-PER
Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology,
Inc., Rockford, IL USA).
Reverse transcription-quantitative
polymerase chain reaction (PCR) analysis
Total RNA was isolated from the cells using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol, and was quantified
spectrophotometrically at 260 nm (ND-1000; Thermo Fisher
Scientific, Inc.). Total RNA (1 μg) was reverse transcribed
using the High Capacity RNA-to-cDNA kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
cDNA was amplified using the SYBR Premix Ex Taq kit (Takara
Bio. Inc., Shiga, Japan) on a StepOnePlus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Briefly, each
reaction volume contained 10 μl SYBR Green PCR Master Mix,
0.8 μM each primer and diethyl pyrocarbonate-treated water,
with a final reaction volume of 20 μl. The primer sequences
were designed using PrimerQuest (Integrated DNA Technologies,
Coralville, IA, USA). The primer sequences were as follows: HO-1,
forward 5′-CTC TTG GCT GGC TTC CTT-3′, reverse 5′-GGC TCC TTC CTC
CTT TCC-3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH),
forward 5′-ACT TTG GTA TCG TGG AAG GACT-3′ and reverse 5′-GTA GAG
GCA GGG ATG ATG TTCT-3. The optimal conditions for PCR
amplification were established according to the manufacturer's
protocol. The thermal cycling conditions used were as follows:
Pre-denaturation at 95°C for 10 min; denaturation at 95°C for 15
sec; and annealing at 60°C for 1 min. A total of 40 cycles were
performed. The data were analyzed using StepOne software (version
2.3; Applied Biosystems; Thermo Fisher Scientific, Inc.), and the
cycle number at the linear amplification threshold (quantification
cycle; Cq) was recorded for the endogenous control gene and the
target gene. Relative gene expression (target gene expression
normalized to the expression of the endogenous control gene) was
calculated using the comparative Cq method (2−ΔΔCq)
(27).
Statistical analysis
The data are presented as the mean ± standard
deviation of at least three independent experiments. To compare
three or more groups, one-way analysis of variance followed by the
Newman-Keuls post-hoc test was conducted. Statistical analysis was
performed using GraphPad Prism software, version 3.03 (GraphPad
Software, Inc., San Diego, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of KCHO-1, glutamate and
H2O2 on HT22 cell viability
The present study examined KCHO-1 cytotoxicity on
HT22 cells using the MTT method. As shown in Fig. 1A, HT22 cells were incubated with
10–400 μg/ml KCHO-1, and cell viability was unchanged
following treatment with all doses of KCHO-1. Therefore, in the
present study, KCHO-1 was used at a concentration of 10–200
μg/ml. Subsequently, the effects of glutamate (0.5–20 mM)
and H2O2 (10–500 μM) were determined
on the viability of HT22 cells. Glutamate significantly reduced
cell viability when used at a concentration of >2 mM (Fig. 1B). H2O2
induced cell death when used at a concentration of >50 μM
(Fig. 1C). Therefore, glutamate
and H2O2 were subsequently used at
concentrations of 5 mM and 100 μM, respectively.
Effects of KCHO-1 on glutamate-induced
oxidative neurotoxicity and ROS generation in HT22 cells
The present study investigated whether KCHO-1
affected glutamate-induced oxidative cell toxicity and ROS
generation in HT22 cells. Cell viability was lower in the
glutamate-treated cells compared with in the control group, whereas
pretreatment with KCHO-1 (100–200 μg/ml) increased viability
in a dose-dependent manner (Fig.
2A). In addition, glutamate treatment doubled ROS production,
whereas KCHO-1 markedly attenuated this increase (Fig. 2B). The known antioxidant trolox was
used as a positive control.
Effects of KCHO-1 on
H2O2-induced oxidative neurotoxicity and ROS
generation in HT22 cells
The present study also determined the protective
action of KCHO-1 against H2O2-induced
neurotoxicity in HT22 cells. Compared with the untreated cells,
treatment with H2O2 caused cell death and
induced ROS production; however, pretreatment with KCHO-1 (100–200
μg/ml) increased viability in a concentration-dependent
manner (Fig. 3A). Furthermore,
KCHO-1 significantly suppressed H2O2-induced
ROS generation (Fig. 3B).
Effects of KCHO-1 on the mRNA and protein
expression levels of HO-1 in HT22 cells
The present study detected HO-1 expression in
KCHO-1-treated HT22 cells. The HT22 cells were treated with
non-cytotoxic concentrations of KCHO-1 (10–200 μg/ml) for 12
h, and HO-1 mRNA (Fig. 4A) and
protein expression levels (Fig.
4B) were increased in a dose-dependent manner. As a HO-1
inducer, CoPP was used as a positive control and dose-dependently
increased HO-1 mRNA and protein expression levels (Fig. 4A and B).
Effects of KCHO-1 on Nrf2 nuclear
translocation in HT22 cells
Nrf2 nuclear translocation is a key inducer of HO-1
expression; therefore, the present study investigated whether
pretreatment of HT22 cells with KCHO-1 also upregulated Nrf2
nuclear translocation (Fig. 5A and
B). Cells were treated with KCHO-1 for 0.5, 1.0 or 1.5 h at a
concentration of 200 μg/ml. Nrf2 levels gradually decreased
in the cytoplasm of HT22 cells (Fig.
5A), whereas nuclear Nrf2 levels markedly increased in a
time-dependent manner (Fig.
5B).
Effects of KCHO-1-induced HO-1 expression
via Nrf2 nuclear translocation on glutamate- and
H2O2-induced oxidative neurotoxicity
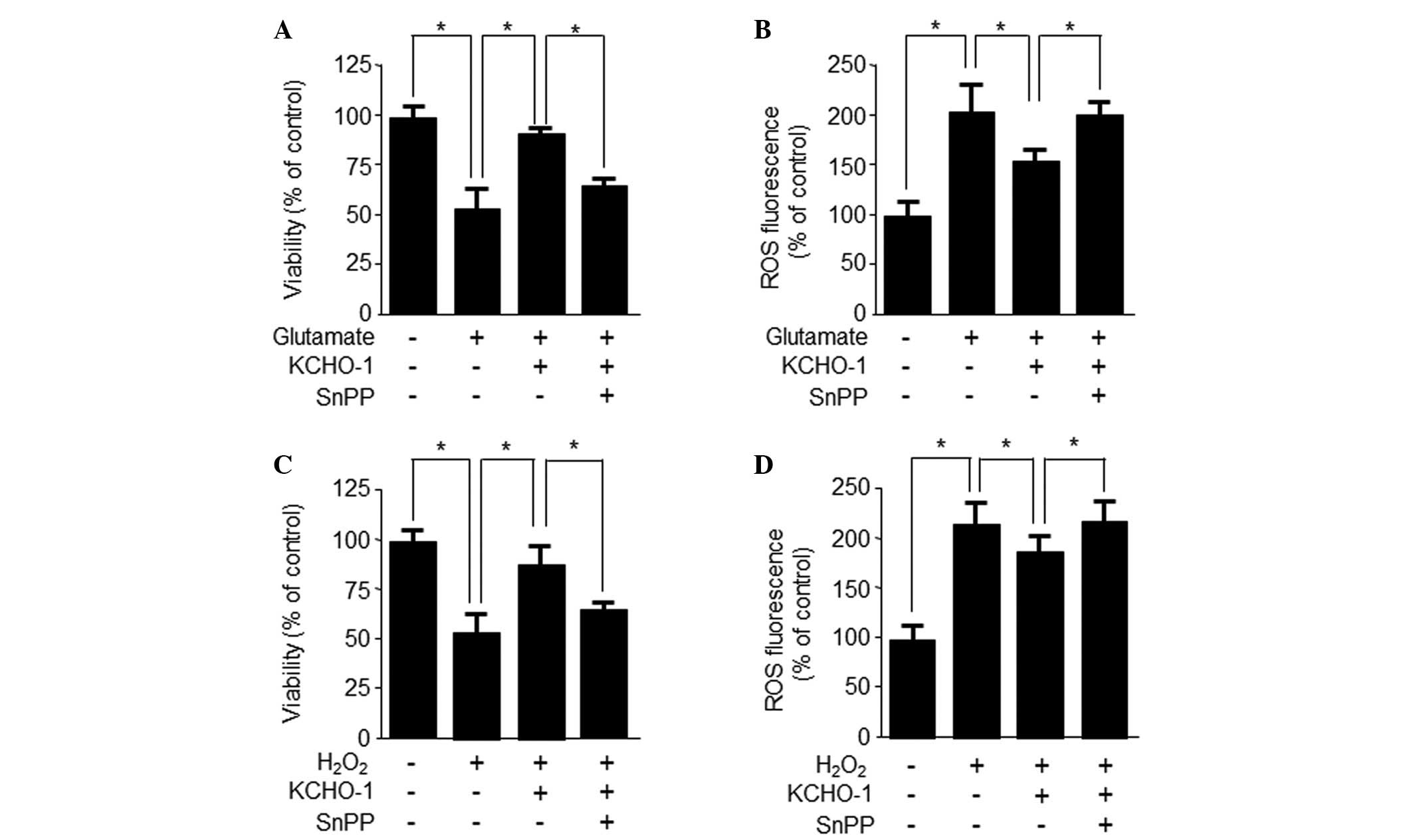
The present study subsequently assessed whether
KCHO-1-induced HO-1 upregulation was responsible for the observed
cytoprotective effects. HT22 cells were co-treated with 200
μg/ml KCHO-1 for 12 h in the absence or presence of the HO
inhibitor SnPP IX. SnPP partially inhibited the ability of KCHO-1
to suppress glutamate-induced cytotoxicity and ROS generation
(Fig. 6A and B). Furthermore, SnPP
partially inhibited the ability of KCHO-1 to suppress
H2O2-induced cytotoxicity and ROS generation
(Fig. 6C and D). These results
suggest that HO-1 expression may be required for the inhibition of
H2O2-induced ROS generation.
Effects of KCHO-1-induced ERK activation
on HO-1 expression, and glutamate- and
H2O2-induced neurotoxicity
To investigate the role of MAPKs in KCHO-1-induced
HO-1 expression, the present study examined the effects of specific
inhibitors, including PD98059 (ERK inhibitor), SP600125 (JNK
inhibitor) and SB203580 (p38 inhibitor). As shown in Fig. 7A, ERK inhibition suppressed
KCHO-1-induced HO-1 expression, whereas JNK and p38 inhibition did
not. In addition, ERK phosphorylation was detected following KCHO-1
treatment between 15 and 60 min (Fig.
7B). PD98059 also partially reversed the ability of KCHO-1 to
inhibit glutamate- and H2O2-induced cell
toxicity (Fig. 7C and D). Data
from the HPLC analysis of KCHO-1 was obtained in the form of
chromatograms by monitoring responses at 254 nm. As presented in
Fig. 8, the retention time of the
main peak was 38.858 min.
Discussion
Oxidative stress in brain tissue may occur
physiologically, as a result of neurodegenerative disorders
(28). Therefore, the authors of
the present study have focused on the mechanism of action of
natural products against neurodegenerative diseases via HO-1
regulation (29–32). In our previous study, the extract
KCHO-1 was developed (26). The
present study investigated the association of HO-1 with the
neuroprotective action of KCHO-1, via Nrf2 nuclear translocation.
To determine the therapeutic potential of KCHO-1, its direct
neuroprotective effects on glutamate- and
H2O2-induced oxidative damage were
investigated in HT22 mouse hippocampal cells.
The HT22 immortalized neuronal cell line has been
used as an in vitro model for mechanistic identification of
glutamate-induced oxidative damage. In the central nervous system,
glutamate is the main excitatory neurotransmitter that is released
by nerve cells in the brain; however, glutamate toxicity induces
neuronal cell death, which is associated with acute insults and
chronic neurodegenerative disorders (33,34).
Glutamate-mediated oxidative stress is caused by inhibiting
cellular cystine uptake, leading to glutathione depletion or ROS
generation and elevated Ca2+ levels (35). H2O2 is the
product of a non-radical two-electron reduction of oxygen, and has
been reported to have a key role in oxidative cell death (6). Therefore, it may be therapeutically
beneficial to reduce the damaging effects of oxidative glutamate or
H2O2 toxicity. As shown in Fig. 1, the present study initially
evaluated the action of glutamate (5 mM) and
H2O2 (100 μM) on the viability of HT22
cells. Subsequently, it was investigated whether KCHO-1 was able to
affect glutamate- or H2O2-induced oxidative
neurotoxicity and ROS generation in HT22 cells. KCHO-1
significantly suppressed glutamate- and
H2O2-induced cell damage and ROS generation
(Figs. 2 and 3).
In our previous studies, it was demonstrated that
HO-1 expression may have an important role in the protection of
HT22 cells (36,37). It has been suggested that the role
of HO-1 in heme degradation may offer cells protection against
oxidative insults and maintain cellular homeostasis. The
antioxidant activities of HO-1 have been observed in Alzheimer's
disease, sepsis, endotoxemia, surgical stress, ischemia reperfusion
injury and psychological stress (26,38).
In the present study, cells were treated with non-cytotoxic
concentrations of KCHO-1. The results indicated that the mRNA and
protein expression levels of HO-1 were increased in HT22 cells
(Fig. 4). Furthermore, the present
study assessed whether KCHO-1-mediated HO-1 upregulation was
responsible for its protective effects on HT22 cells. Treatment
with the HO-1 inhibitor SnPP partially reversed the ability of
KCHO-1 to inhibit H2O2-induced cell death and
ROS generation (Fig. 6). These
results suggested that HO-1 expression may be required to inhibit
H2O2-induced ROS generation. Nrf2 is a basic
leucine zipper transcription factor, which resides in the cytoplasm
bound to Keap-1. Following stimulation with inducers, Nrf2
translocates into the nucleus (39–41).
Nrf2 has been reported to induce the expression of antioxidant
proteins, including HO-1 (42).
The present study revealed that KCHO-1 significantly upregulated
Nrf2 and efficiently promoted its translocation into the nucleus,
thus suggesting that KCHO-1-induced HO-1 expression may be
associated with Nrf2 nuclear translocation (Fig. 5).
The present study also demonstrated that the ERK
pathway is involved in KCHO-1-induced HO-1 expression (Fig. 7). MAPK is one of the most common
cellular response signaling pathways, which responds to various
extracellular stimuli. There are three subfamilies of MAPK: p38
kinase, ERK1/2 and JNK (43).
MAPKs are initiated in response to various extracellular stimuli,
particularly oxidative stress. Previous studies have reported that
activation of MAPK pathways may contribute to HO-1 gene expression
(44,45). In the present study, KCHO-1-induced
HO-1 gene expression was shown to be associated with the ERK
pathway, since treatment with the ERK inhibitor, PD98059,
suppressed KCHO-1-induced HO-1 expression; however, JNK and p38
inhibition did not affect HO-1 expression. As expected, treatment
with the ERK pathway inhibitor also abolished KCHO-1-induced
cytoprotection (Fig. 7). These
results indicated that KCHO-1-induced HO-1 expression in HT22 cells
may be mediated by the Nrf2 or ERK pathways.
In conclusion, the results of the present study
suggested that KCHO-1 may effectively prevent glutamate- or
H2O2-induced oxidative cell damage in a
murine hippocampal cell line. KCHO-1-induced HO-1 upregulation via
ERK and Nrf2 pathways appears to have a central role in the
protection of HT22 cells. These results may provide an insight into
the mechanisms underlying KCHO-1-induced neuronal cell protection
and HO-1 enzyme induction. Therefore, KCHO-1 may be considered a
potential agent for the treatment of neurodegenerative
diseases.
Acknowledgments
The present study was supported by the Traditional
Korean Medicine R&D Program funded by the Ministry of Health
& Welfare through the Korea Health Industry Development
Institute (KHIDI) (grant no. HI11C2142).
References
|
1
|
Chen YR and Zweier JL: Cardiac
mitochondria and reactive oxygen species generation. Circ Res.
114:524–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paulsen JS, Nance M, Kim JI, Carlozzi NE,
Panegyres PK, Erwin C, Goh A, McCusker E and Williams JK: A review
of quality of life after predictive testing for and earlier
identification of neurodegenerative diseases. Prog Neurobiol.
110:2–28. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sena LA and Chandel NS: Physiological
roles of mitochondrial reactive oxygen species. Mol Cell.
48:158–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dobrek L and Thor P: Glutamate NMDA
receptors in pathophysiology and pharmacotherapy of selected
nervous system diseases. Postepy Hig Med Dosw (Online). 65:338–346.
2011. View Article : Google Scholar
|
|
6
|
Sies H: Role of metabolic
H2O2 generation: Redox signaling and
oxidative stress. J Biol Chem. 289:8735–8741. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Girvan HM and Munro AW: Heme sensor
proteins. J Biol Chem. 288:13194–13203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang X, Cao J, Sun BW, Liu DD, Liang F and
Gao L: Exogenous carbon monoxide attenuates inflammatory responses
in the small intestine of septic mice. World J Gastroenterol.
18:5719–5728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ryter SW and Choi AM: Heme
oxygenase-1/carbon monoxide: From metabolism to molecular therapy.
Am J Respir Cell Mol Biol. 41:251–260. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Al-Owais MM, Scragg JL, Dallas ML, Boycott
HE, Warburton P, Chakrabarty A, Boyle JP and Peers C: Carbon
monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in
medulloblastoma DAOY cells via K+ channel inhibition. J
Biol Chem. 287:24754–24764. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Parfenova H, Leffler CW, Basuroy S, Liu J
and Fedinec AL: Antioxidant roles of heme oxygenase, carbon
monoxide, and bilirubin in cerebral circulation during seizures. J
Cereb Blood Flow Metab. 32:1024–1034. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jansen T, Hortmann M, Oelze M, Opitz B,
Steven S, Schell R, Knorr M, Karbach S, Schuhmacher S, Wenzel P, et
al: Conversion of biliverdin to bilirubin by biliverdin reductase
contributes to endothelial cell protection by heme
oxygenase-1-evidence for direct and indirect antioxidant actions of
bilirubin. J Mol Cell Cardiol. 49:186–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lipiński P, Jarzabek Z, Broniek S and
Zagulski T: Protective effect of tissue ferritins in experimental
Escherichia coli infection of mice in vivo. Int J Exp Pathol.
72:623–630. 1991.
|
|
14
|
Chen B, Lu Y, Chen Y and Cheng J: The role
of Nrf2 in oxidative stress-induced endothelial injuries. J
Endocrinol. 225:R83–R99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Farombi EO and Surh YJ: Heme oxygenase-1
as a potential therapeutic target for hepatoprotection. J Biochem
Mol Biol. 39:479–491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim EK and Choi EJ: Pathological roles of
MAPK signaling pathways in human diseases. Biochim Biophys Acta.
1802:396–405. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang LH, Li Y, Yang SN, Wang FY, Hou Y,
Cui W, Chen K, Cao Q, Wang S, Zhang TY, et al: Gambogic acid
synergistically potentiates cisplatin-induced apoptosis in
non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1
signalling. Br J Cancer. 110:341–352. 2014. View Article : Google Scholar :
|
|
18
|
Lee DS, Kim KS, Ko W, Li B, Jeong GS, Jang
JH, Oh H and Kim YC: The cytoprotective effect of sulfuretin
against tert-butyl hydroperoxide-induced hepatotoxicity through
Nrf2/ARE and JNK/ERK MAPK-mediated heme oxygenase-1 expression. Int
J Mol Sci. 15:8863–8877. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ringman JM, Frautschy SA, Cole GM,
Masterman DL and Cummings JL: A potential role of the curry spice
curcumin in Alzheimer's disease. Curr Alzheimer Res. 2:131–136.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang GB, Zhao T, Muna SS, Jin HM, Park
JI, Jo KS, Lee BH, Chae SW, Kim SY, Park SH, et al: Therapeutic
potential of Gastrodia elata Blume for the treatment of Alzheimer's
disease. Neural Regen Res. 8:1061–1070. 2013.PubMed/NCBI
|
|
21
|
Li Z, Liu Y, Wang L, Liu X, Chang Q, Guo
Z, Liao Y, Pan R and Fan TP: Memory-enhancing effects of the crude
extract of Polygala tenuifolia on aged mice. Evid Based Complement
Alternat Med. 2014:3923242014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dittmann K, Gerhäuser Klimo CK and
Hamburger M: HPLC-based activity profiling of Salvia miltiorrhiza
for MAO A and iNOS inhibitory activities. Planta Med. 70:909–913.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Han YJ, Je JH, Kim SH, Ahn SM, Kim HN, Kim
YR, Choi YW, Shin HK and Choi BT: Gastrodia elata shows
neuroprotective effects via activation of PI3K signaling against
oxidative glutamate toxicity in HT22 cells. Am J Chin Med.
42:1007–1019. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu Y, Liu M, Liu P, Guo DH, Wei RB and
Rahman K: Possible mechanism of the antidepressant effect of
3,6′-disinapoyl sucrose from Polygala tenuifolia Willd. J Pharm
Pharmacol. 63:869–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hwang CK and Chun HS: Isoliquiritigenin
isolated from licorice Glycyrrhiza uralensis prevents
6-hydroxydopamine-induced apoptosis in dopaminergic neurons. Biosci
Biotechnol Biochem. 76:536–543. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee DS, Ko W, Yoon CS, Kim DC, Yun J, Lee
JK, Jun KY, Son I, Kim DW, Song BK, et al: KCHO-1, a novel
antineuroinflammatory agent, inhibits lipopolysaccharide-induced
neuroinflammatory responses through Nrf2-mediated heme oxygenase-1
expression in mouse BV2 microglia cells. Evid Based Complement
Alternat Med. 2014:3571542014. View Article : Google Scholar
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
28
|
Shimomura H, Ogawa H, Takazoe K, Soejima
H, Miyamoto S, Sakamoto T, Kawano H, Suefuji H, Nishikawa H, Arai
H, et al: Comparison of urinary biopyrrin levels in acute
myocardial infraction (after reperfusion therapy) versus stable
angina pectoris and their usefulness in predicting subsequent
cardiac events. Am J Cardiol. 90:108–111. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hald A and Lotharius J: Oxidative stress
and inflammation in Parkinson's disease: Is there a causal link?
Exp Neurol. 193:279–290. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee DS, Ko W, Quang TH, Kim KS, Sohn JH,
Jang JH, Ahn JS, Kim YC and Oh H: Penicillinolide A: A new
anti-inflammatory metabolite from the marine fungus Penicillium sp
SF-5292. Mar Drugs. 11:4510–4526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee DS, Kim KS, Ko W, Li B, Keo S, Jeong
GS, Oh H and Kim YC: The neoflavonoid latifolin isolated from MeOH
extract of Dalbergia odorifera attenuates inflammatory responses by
inhibiting NF-κB activation via Nrf2-mediated heme oxygenase-1
expression. Phytother Res. 28:1216–1223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee DS, Li B, Im NK, Kim YC and Jeong GS:
4,2′,5′-trihydroxy-4′- methoxychalcone from Dalbergia odorifera
exhibits anti-inflammatory properties by inducing heme oxygenase-1
in murine macrophages. Int Immunopharmacol. 16:114–121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Keller JN and Mattson MP: Roles of lipid
peroxidation in modulation of cellular signaling pathways, cell
dysfunction and death in the nervous system. Rev Neurosci.
9:105–116. 1998. View Article : Google Scholar
|
|
34
|
Siesjö BK: Cell damage in the brain: A
speculative synthesis. J Cereb Blood Flow Metab. 1:155–185. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mattson MP: Apoptosis in neurodegenerative
disorders. Nat Rev Mol Cell Biol. 1:120–129. 2000. View Article : Google Scholar
|
|
36
|
Lee DS and Jeong GS: Arylbenzofuran
isolated from Dalbergia odorifera suppresses
lipopolysaccharide-induced mou s e BV2 microglial cell activation,
which protects mouse hippocampal HT22 cells death from
neuroinflammation-mediated toxicity. Eur J Pharmacol. 728:1–8.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee DS, Ko W, Kim DC, Kim YC and Jeong GS:
Cudarflavone B provides neuroprotection against glutamate-induced
mouse hippocampal HT22 cell damage through the Nrf2 and PI3K/Akt
signaling pathways. Molecules. 19:10818–10831. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamaguchi T, Hasizume T, Tanaka M,
Nakayama M, Sugimoto A, Ikeda S, Nakajima H and Horio F: Bilirubin
oxidation provoked by endotoxin treatment is suppressed by feeding
ascorbic acid in a rat mutant unable to synthesize ascorbic acid.
Eur J Biochem. 245:233–240. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee BS, Heo J, Kim YM, Shim SM, Pae HO,
Kim YM and Chung HT: Carbon monoxide mediates heme oxygenase 1
induction via Nrf2 activation in hepatoma cells. Biochem Biophys
Res Commun. 343:965–972. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qiang W, Cahill JM, Liu J, Kuang X, Liu N,
Scofield VL, Voorhees JR, Reid AJ, Yan M, Lynn WS and Wong PK:
Activation of transcription factor Nrf-2 and its downstream targets
in response to moloney murine leukemia virus ts1-induced thiol
depletion and oxidative stress in astrocytes. J Virol.
78:11926–11938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim KM, Pae HO, Zheng M, Park R, Kim YM
and Chung HT: Carbon monoxide induces heme oxygenase-1 via
activation of protein kinase R-like endoplasmic reticulum kinase
and inhibits endothelial cell apoptosis triggered by endoplasmic
reticulum stress. Circ Res. 101:919–927. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lim HJ, Lee KS, Lee S, Park JH, Choi HE,
Go SH, Kwak HJ and Park HY: 15d-PGJ2 stimulates HO-1 expression
through p38 MAP kinase and Nrf-2 pathway in rat vascular smooth
muscle cells. Toxicol Appl Pharmacol. 223:20–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Choi BH, Hur EM, Lee JH, Jun DJ and Kim
KT: Protein kinase Cdelta-mediated proteasomal degradation of MAP
kinase phosphatase-1 contributes to glutamate-induced neuronal cell
death. J Cell Sci. 119:1329–1340. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Satoh T, Nakatsuka D, Watanabe Y, Nagata
I, Kikuchi H and Namura S: Neuroprotection by MAPK/ERK kinase
inhibition with U0126 against oxidative stress in a mouse neuronal
cell line and rat primary cultured cortical neurons. Neurosci Lett.
288:163–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Elbirt KK, Whitmarsh AJ, Davis RJ and
Bonkovsky HL: Mechanism of sodium arsenite-mediated induction of
heme oxygenase-1 in hepatoma cells. Role of mitogen-activated
protein kinases. J Biol Chem. 273:8922–8931. 1998. View Article : Google Scholar : PubMed/NCBI
|