Introduction
Viral myocarditis is a potentially life-threatening
disease. It often results in heart failure and mortality due to
cardiac contractile dysfunction, characterized by systolic and
diastolic dysfunction (1,2). As a member of the positive-stranded
RNA virus family, Picornaviridae, coxsackie virus B3 (CVB3) has
been confirmed to be one of the major viruses identified to cause
acute viral myocarditis (3).
Cardiac fibroblasts, comprising up to 65–70% of the cells in the
myocardium, are important in the pathology of CVB3-induced
myocarditis (4,5). It has been reported that CVB3
infection can cause interstitial collagen deposition resulting in
cardiac fibrosis (6). The
inhibition of cardiac fibroblast formation and collagen synthesis
can provide a means of attenuating and preventing cardiac fibrosis
and its sequelae (7).
Adenosine monophosphate-activated protein kinase
(AMPK) has been considered as a cellular fuel gauge and super
metabolic regulator (8). Its
protective role during myocardial ischemia has been reported in
detail (9), however, whether AMPK
has an effect on the inflammatory cardiomyopathy and myocardial
fibrosis induced by CVB3 remains to be elucidated. A previous study
reported an association between AMPK and CVB3 replication;
activation of AMPK was shown to restrict CVB3 replication by
inhibiting lipid accumulation (10). Therefore, the present study aimed
to investigate the role of AMPK activation in collagen production
in CVB3-infected cardiac fibroblasts, as well as the underlying
mechanism.
Materials and methods
Reagents
5-aminoimidazole-4-carboxamide-ribonucleoside
(AICAR) was purchased from Toronto Research Chemicals (Toronto, ON,
Canada). Compound C was purchased from EMD Biosciences (San Diego,
CA, USA), and SB203580 was purchased from Sigma-Aldrich (St. Louis,
MO, USA). Rabbit anti-phosphorylated (p)-AMPKα-Thr172
(1:1,000; cat. no. 2531), anti-AMPKα (1:1,000; cat. no. 2532),
anti-p-p38 (1:1,000; cat. no. 9211) and anti-p38 (1:1,000; cat. no.
9212) polyclonal antibodies were obtained from Cell Signaling
Technology, Inc. (Beverly, MA, USA). Mouse anti-collagen I
monoclonal antibody (1:200; cat. no. sc-59772), goat anti-collagen
IV polyclonal antibody (1:200; cat. no. sc-9301), rabbit
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) monoclonal
antibody (1:3,000; cat. no. sc-32233) and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit (1:6,000; cat. no. sc-2004) and
goat anti-mouse (1:6,000; cat. no. sc-2005) secondary antibodies
were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Fetal bovine serum (FBS) was obtained from GE Healthcare Life
Sciences (Logan, UT, USA).
Determination of viral titers
HeLa cells [CCL-2; American Type Culture Collection
(ATCC), Manassas, VA, USA] were used to propagate the CVB3 Nancy
strain (ATCC). Firstly, HeLa cells grown to 80% confluency were
washed two times in 10 ml 1X phosphate-buffered saline (PBS), then
CVB3 solution was added to HeLa cells and incubated at 37°C in 5%
CO2 for 1 h. Dulbecco's modified Eagle's medium (DMEM;
GE Healthcare Life Sciences) supplemented with 10% FBS was added
and the plate was incubated at 37°C in 5% CO2 for an
additional 48 h. Subsequently, the infected HeLa cells were lysed
by three cycles of freezing (−80°C) and thawing (37°C). Following
centrifugation at 3,000 × g for 10 min at room temperature, the
debris was removed and serial dilutions (10-fold;
1×10−1–10−9) of the supernatants were
prepared using infection medium containing minimal essential medium
(GE Healthcare Life Sciences), 2% FBS, 30 mM MgCl2, 100
U/ml penicillin, 100 µg/ml streptomycin sulfate and 2 mM
glutamine. Subsequently, the serial supernatant dilutions were
transferred onto subconfluent monolayers of HeLa cells grown in
96-well culture plates containing 100 µl infection medium.
Following incubation at 37°C for 24 h, the cells were stained with
0.5% crystal violet (in water) for 5 min and visualized using an
inverted microscope (Olympus IX73; Olympus Corporation, Tokyo,
Japan). The 50% tissue culture infectious dose (TCID50) was
calculated using the Reed-Muench method (11).
Cell isolation and culture
The cultured neonatal rat cardiac fibroblasts
(NRCFs) were isolated from 1-day-old Sprague-Dawley rats. Briefly,
following anesthesia with 2% ether, the chest cavities of the
neonatal rats were opened and the rat hearts were harvested.
Subsequently, the neonatal rat hearts were minced and digested
using 0.1% trypsin (Roche Diagnostics GmbH, Mannheim, Germany) and
0.03% collagenase II (Worthington Biochemical Corporation,
Lakewood, NJ, USA). The collected cells were seeded into 10-cm cell
culture plates containing 10 ml DMEM supplemented with 1%
penicillin-streptomycin and 10% FBS. In order to allow the
fibroblasts to attach to the cell culture plates, the cell
suspension was placed in a 37°C 5% CO2 incubator for 60
min. Subsequently, the fibroblasts were washed twice with DMEM and
cultured in DMEM with 10% FBS at 37°C for 48 h until they reached
confluence. All the NRCFs were incubated in DMEM with 2% FBS for 24
h prior to performing the respective experiments. The animal care
and experimental protocols were in compliance with the Animal
Management Rule of the People's Republic of China (The State
Council of the people's Republic of China; http://www.gov.cn/gongbao/content/2014/content_2692743.htm).
The present study was approved by the Committee on Ethics of
Biomedicine Research at the Third Affiliated Hospital of Nantong
University (Wuxi, China).
Hydroxyproline measurement
The cell culture supernatants were collected
following treatment, following which the levels of hydroxproline
were detected using a hydroxyproline ELISA kit (cat. no. KB2847A;
Kaibo Biochemical Reagents Co., Ltd., Shanghai, China), according
to the manufacturer's protocol. Hydroxyproline concentrations were
determined by the optical density values at 450 nm on an ELISA
plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Western blot analysis
Following washing once with cold PBS, the cell
samples were lysed in buffer containing 10 mmol/l sodium
pyrophosphate, 100 mmol/l NaCl, 50 mmol/l NaF, 1 mmol/l sodium
vanadate, 5 mmol/l EDTA, 1% sodium deoxycholate, 20 mmol/l Tris-HCl
(pH 7.4), 0.1% sodium dodecyl sulphate (SDS), 1 mmol/l
phenylmethylsulphonyl fluoride, 0.1 mmol/l aprotinin, 1 mmol/l
leupeptin, 1% Triton X-100 and 10% glycerol. The protein
concentrations were estimated using a bicinchoninic acid protein
assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). Equal
quantities of the protein samples (30 µg) were mixed with an
equal volume of 2X SDS sample buffer containing 4% SDS, 100 mM
Tris-HCl (pH 6.8), 20% glycerol, 200 mM DTT and 0.2% bromophenol
blue. After boiling the mixture at 95°C for 5 min, the protein
samples were loaded and separated on 8 or 10% SDS-polyacrylamide
gels. Subsequently, the samples were electroblotted onto
nitrocellulose membranes (Pall Life Sciences, East Hill, NY, USA).
Nonspecific binding was then blocked with 5% nonfat milk. The
membranes were incubated overnight at 4°C with rabbit
anti-p-AMPKα-Thr172 (1:1,000), anti-AMPKα (1:1,000),
anti-p-p38 (1:1,000) and anti-p38 (1:1,000) polyclonal antibodies,
mouse anti-collagen I monoclonal antibody (1:200), goat
anti-collagen IV polyclonal antibody (1:200) and rabbit anti-GAPDH
monoclonal antibody (1:3,000), followed by incubation with
HRP-conjugated goat anti-rabbit and anti-mouse secondary antibodies
(1:6,000) for 1 h at room temperature. An enhanced
chemiluminescence system (ECL kit; Pierce Biotechnology, Inc.) was
used to visualize the membranes. The following primary antibodies
were used, at 1:1,000 dilutions: Anti-collagen I, anti-collagen IV,
anti-AMPKα, anti-p-AMPKα (Thr172), anti-p38, anti-p-p38
and anti-GAPDH.
Statistical analysis
All the experiments were performed at least three
times. Data are expressed as the mean ± standard error of the mean.
One-way analysis of variance was performed for multiple-group
comparisons, and unpaired two-tailed t-tests were used to
evaluate the significance between groups. Statistical analyses were
performed using SPSS software 20.0 (IBM SPSS, Armonk, NY, USA).
P<0.05 were considered to indicate a statistically significant
difference.
Results
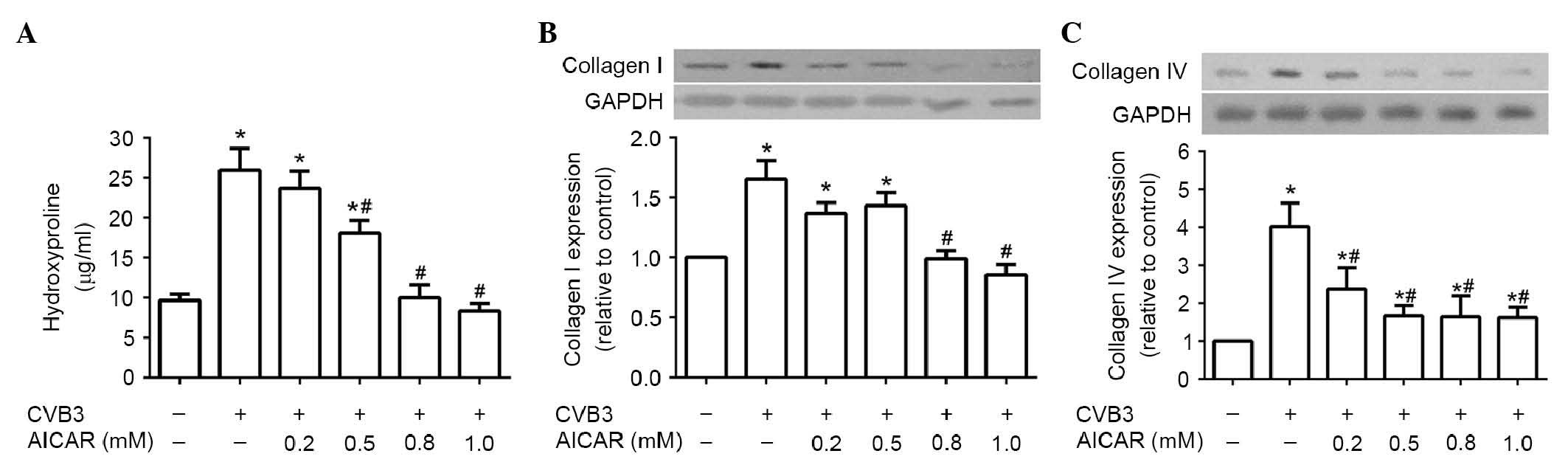
AICAR reduces collagen production in
CVB3-induced NRCFs in a dose-dependent manner
Although a substantial volume of data regarding AMPK
and collagen secretion is now available, their association in NRCFs
induced by CVB3 remains to be fully elucidated. In the present
study, it was hypothesized that the activation of AMPK may affect
the production of collagen in the NRCFs infected with CVB3.
Following treatment with AICAR for 30 min, 5×104 NRCFs
incubated in DMEM with 2% FBS were infected with CVB3 (100 TCID50)
for 48 h. As expected, CVB3 infection promoted the levels of
hydroxyproline in the supernatant (Fig. 1A) and the production of collagen
I/collagen IV (Fig. 1B and C) in
the cell lysate, compared with the control group. However, these
effects were dose-dependently inhibited following pretreatment with
AICAR.
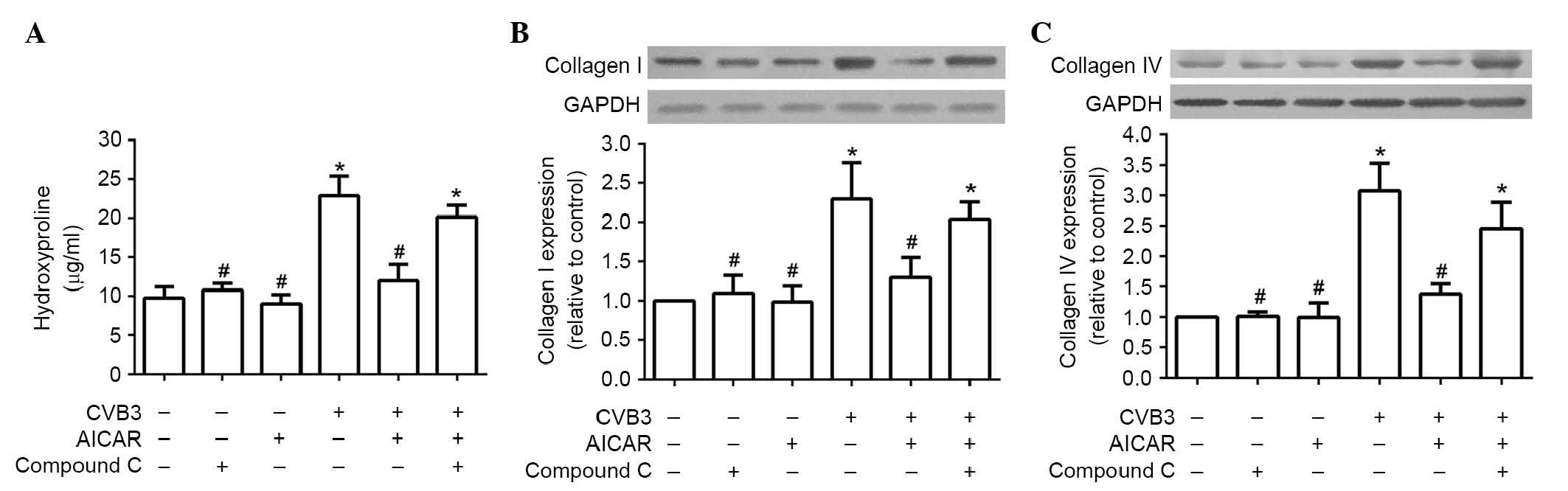
Inhibitory effects of AICAR are reversed
by compound C
To examine whether AMPK is a involved in the
inhibitory effects of AICAR, the cardiac fibroblasts of mice
(5×104 per well) were preincubated with 1 µM
compound C, an AMPK inhibitor, for 30 min, followed by treatment of
the cells with AICAR (1 mM) and CVB3 (100 TICD50), as above. In the
NRCFs preincubated with compound C, return of the CVB3-induced
increases in hydroxyproline content in the supernatant (Fig. 2A) and the production of collagen
I/collagen IV (Fig. 2B and C) in
the cell lysate were observed.
P38 mitogen-activated protein kinase
(MAPK) is a downstream kinase of AMPK in NRCFs
It has been reported that p38 MAPK is an important
regulator in collagen production of fibroblasts (12). Therefore, the present study
examined whether p38 MAPK was involved in the inhibitory effects of
AICAR in the CVB3-infected NRCFs. It was observed that AMPK and p38
phosphorylation were significantly increased by AICAR in the NRCFs
(Fig. 3A). SB03580, a p38
MAPK-specific inhibitor, inhibited p38 MAPK phosphorylation in a
dose-dependent manner, however, AMPK was not affected (Fig. 3B).
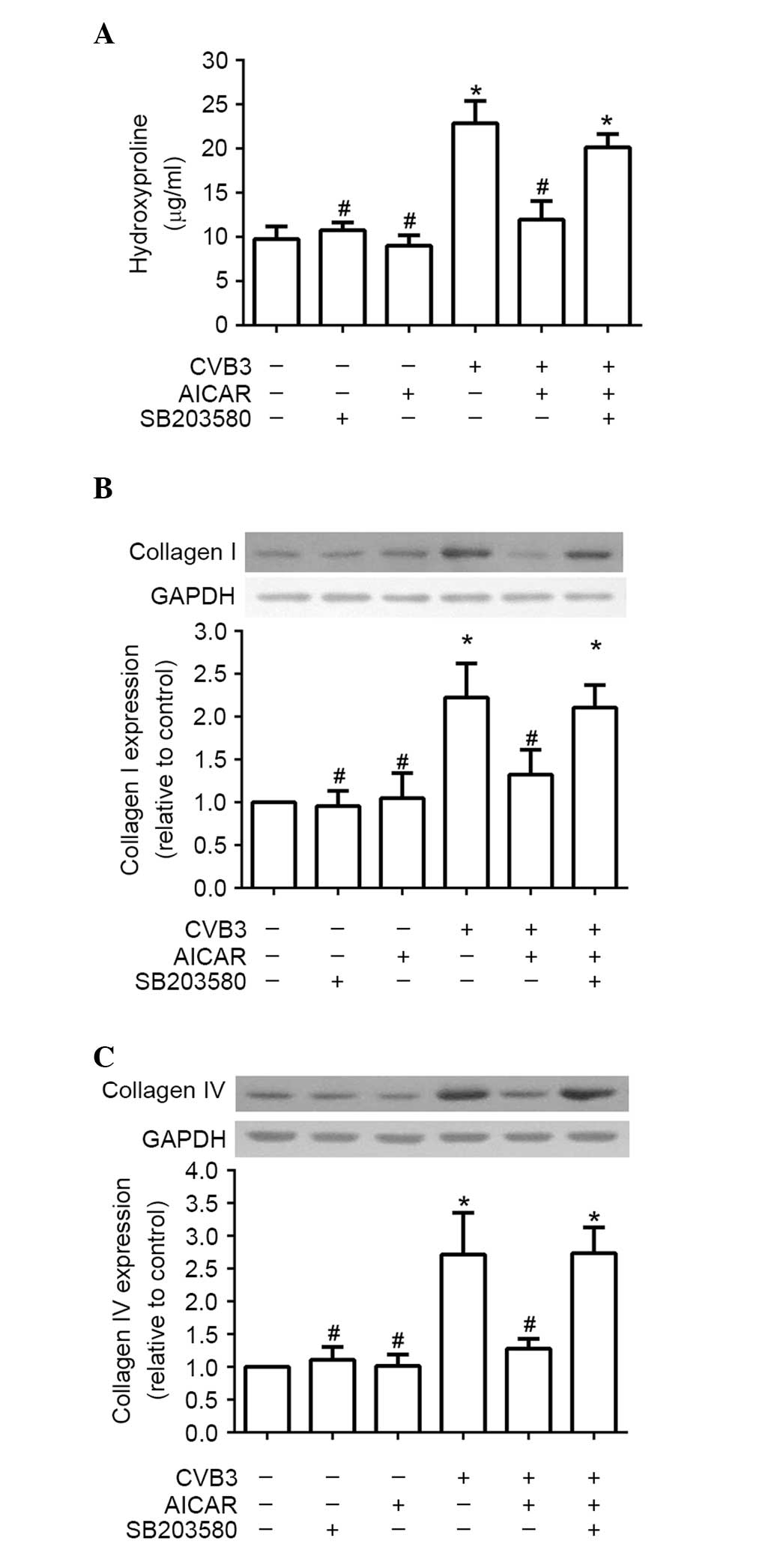
P38 MAPK is required for the inhibitory
effect of AICAR on collagen production of NRCFs induced by
CVB3
To confirm the hypothesis that p38 MAPK is required
for the inhibitory effects of AICAR in the collagen production of
NRCFs induced by CVB3, the NRCFs were pre-incubated with SB203580
(10 µM) for 30 min. The cells were then treated with AICAR
(1 mM) and CVB3 (100 TICD50) in turn, as described above. Similar
effects to those of compound C were observed. The CVB3-induced
increases in hydroxyproline content in the supernatant (Fig. 4A) and production of collagen
I/collagen IV (Fig. 4B and C) in
the cell lysate returned following pretreatment with SB203580.
Discussion
CVB3 infection commonly induces viral myocarditis
and dilated cardiomyopathy, eventually resulting in heart failure
(13). Interstitial collagen
deposition and the fibrotic remodelling of cardiac tissues are key
determinants in the progression of heart failure (14). Cardiac fibroblasts, which produce
and degrade the myocardial extracellular matrix, are critical in
cardiac inflammation and maladaptive ventricular remodeling.
Cardiac fibroblasts are involved in the pathology of CVB3-induced
myocarditis and dilated cardiomyopathy by promoting cell-specific
viral replication (5), aggravating
collagen deposition (15) and
immunolesions (16).
Hydroxyproline, a major component of collagen, can stabilize the
triple-helical structure of collagen via maximizing interchain
hydrogen bond formation (17). The
measurement of hydroxyproline levels has been used as an indicator
of collagen content (18). In the
present study, it was observed that CVB3 infection promoted
hydroxyproline content in the supernatant and the production of
collagen I/collagen IV in the cell lysate, compared with the
control group.
AMPK, a heterotrimeric complex with a catalytic (α)
and two regulatory subunits (β and γ) has been confirmed as a key
regulator of cellular energy homeostasis (19). It monitors the AMP/ATP ratio to
regulate cellular metabolism by restoring levels of ATP. The
α-subunit, containing a serine-threonine kinase domain, is
essential for AMPK activity. It has a critical activating residue
within the catalytic cleft (Thr172) (20,21).
The phosphorylation status of α-Thr172 has been used as
an indicator of the state of AMPK activation (22). AMPK can be activated by a variety
of signals, which are not directly associated with metabolism,
including ischemia (22), hypoxia,
oxidative stress, catecholamines and nitric oxide (23). Furthermore, a variety of signal
transduction proteins, including endothelial nitric oxide synthase
(24), c-Jun N-terminal kinase
(25) and p38MAPK (26), can be activated by AMPK. It has
been reported that AMPK is involved in certain heart diseases,
including myocardial ischemia (22,27),
pressure overload or thyroid hormone-induced cardiac hypertrophy
(28,29) and heart failure (30). However, whether AMPK activation is
involved in viral myocarditis induced by CVB3 remains to be
elucidated.
In the present study, the production of collagen in
NRCFs induced by CVB3 was dose-dependently inhibited by AICAR, a
well-known pharmacological activator of AMPK. Pretreatment with
compound C, an AMPK inhibitor, and SB203580 a p38 inhibitor,
reversed the effects of AICAR. AICAR significantly increased the
levels of AMPKα-Thr172 and p38 MAPK phosphorylation.
However, only p38 MAPK phosphorylation was inhibited by SB203580.
These results indicated that p38 may be the downstream kinase of
AMPK in CVB3-infected NRCFs. The pharmacological activation of AMPK
reduced collagen production via the p38 MAPK-dependent pathway in
the cardiac fibroblasts induced by CVB3. The secretion of collagen
by cardiac fibroblasts is a key cause of myocardial fibrosis
(31). This finding suggests that
AMPK, in addition to its effects on glucose metabolism, may be
involved in the cardiac fibrosis observed in the CVB3-infected
heart.
In conclusion, the present study demonstrated that
the activation of AMPK with AICAR inhibited collagen production via
the p38 MAPK-dependent pathway in CVB3-infected NRCFs. The
inhibitory effects of AMPK activation in the production of collagen
in CVB3-infected cardiac fibroblasts may contribute to identifying
an effective therapy for CVB3-induced myocarditis and
CVB3-associated dilated cardiomyopathy.
Acknowledgments
This study was supported by the Natural Science
Foundation of China (grant no. 81200161) and the Wuxi Hospital
Management Center Project (grant no. YGZXM14012).
References
|
1
|
Ellis CR and Di Salvo T: Myocarditis:
Basic and clinical aspects. Cardiol Rev. 15:170–177. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Magnani JW and Dec GW: Myocarditis:
Current trends in diagnosis and treatment. Circulation.
113:876–890. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sagar S, Liu PP and Cooper LT Jr:
Myocarditis. Lancet. 379:738–747. 2012. View Article : Google Scholar
|
|
4
|
Eghbali M: Cardiac fibroblasts: Function,
regulation of gene expression and phenotypic modulation. Basic Res
Cardiol. 87(Suppl 2): S183–S189. 1992.
|
|
5
|
Lindner D, Li J, Savvatis K, Klingel K,
Blankenberg S, Tschöpe C and Westermann D: Cardiac fibroblasts
aggravate viral myocarditis: Cell specific coxsackievirus B3
replication. Mediators Inflamm. 2014:5195282014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cao Y, Xu W and Xiong S: Adoptive transfer
of regulatory T cells protects against Coxsackievirus B3-induced
cardiac fibrosis. PLoS One. 8:e749552013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Swaney JS, Roth DM, Olson ER, Naugle JE,
Meszaros JG and Insel PA: Inhibition of cardiac myofibroblast
formation and collagen synthesis by activation and overexpression
of adenylyl cyclase. Proc Natl Acad Sci USA. 102:437–442. 2005.
View Article : Google Scholar :
|
|
8
|
Zaha VG and Young LH: AMP-activated
protein kinase regulation and biological actions in the heart. Circ
Res. 111:800–814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Horman S, Beauloye C, Vanoverschelde JL
and Bertrand L: AMP-activated protein kinase in the control of
cardiac metabolism and remodeling. Curr Heart Fail Rep. 9:164–173.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie W, Wang L, Dai Q, Yu H, He X, Xiong J,
Sheng H, Zhang D, Xin R, Qi Y, et al: Activation of AMPK restricts
coxsackievirus B3 replication by inhibiting lipid accumulation. J
Mol Cell Cardiol. 85:155–167. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Reed LJ and Muench H: A simple method of
estimating fifty percent endpoints. Am J Hyg. 27:493–497. 1938.
|
|
12
|
Yu XY, Qiao SB, Guan HS, Liu SW and Meng
XM: Effects of visfatin on proliferation and collagen synthesis in
rat cardiac fibroblasts. Horm Metab Res. 42:507–513. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu PP and Yan AT: Cardiovascular magnetic
resonance for the diagnosis of acute myocarditis: Prospects for
detecting myocardial inflammation. J Am Coll Cardiol. 45:1823–1825.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pchejetski D, Foussal C, Alfarano C,
Lairez O, Calise D, Guilbeau-Frugier C, Schaak S, Seguelas MH,
Wanecq E, Valet P, et al: Apelin prevents cardiac fibroblast
activation and collagen production through inhibition of
sphingosine kinase 1. Eur Heart J. 33:2360–2369. 2012. View Article : Google Scholar
|
|
15
|
Leipner C, Grün K, Müller A, Buchdunger E,
Borsi L, Kosmehl H, Berndt A, Janik T, Uecker A, Kiehntopf M and
Böhmer FD: Imatinib mesylate attenuates fibrosis in coxsackievirus
b3-induced chronic myocarditis. Cardiovasc Res. 79:118–126. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu M, Hu J, Zhu MX, Zhao T, Liang W, Wen
S, Li HH, Long Q, Wang M, Guo HP, et al: Cardiac fibroblasts
recruit Th17 cells infiltration into myocardium by secreting CCL20
in CVB3-induced acute viral myocarditis. Cell Physiol Biochem.
32:1437–1450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bhattacharjee A and Bansal M: Collagen
structure: The Madras triple helix and the current scenario. IUBMB
Life. 57:161–172. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kasyanov V, Moreno-Rodriguez RA, Kalejs M,
Ozolanta I, Stradins P, Wen X, Yao H and Mironov V: Age-related
analysis of structural, biochemical and mechanical properties of
the porcine mitral heart valve leaflets. Connect Tissue Res.
54:394–402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Puthanveetil P, Wang F, Kewalramani G, Kim
MS, Hosseini-Beheshti E, Ng N, Lau W, Pulinilkunnil T, Allard M,
Abrahani A and Rodrigues B: Cardiac glycogen accumulation after
dexamethasone is regulated by AMPK. Am J Physiol Heart Circ
Physiol. 295:H1753–H1762. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu WY and Jiang RS: Advances in the
research of AMPK and its subunit genes. Pak J Biol Sci.
16:1459–1468. 2013. View Article : Google Scholar
|
|
21
|
Xiao B, Sanders MJ, Underwood E, Heath R,
Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, et
al: Structure of mammalian AMPK and its regulation by ADP. Nature.
472:230–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Baron SJ, Li J, Russell RR III, Neumann D,
Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA and Young
LH: Dual mechanisms regulating AMPK kinase action in the ischemic
heart. Circ Res. 96:337–345. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Novikova DS, Garabadzhiu AV, Melino G,
Barlev NA and Tribulovich VG: AMP-activated protein kinase:
Structure, function, and role in pathological processes.
Biochemistry (Mosc). 80:127–144. 2015. View Article : Google Scholar
|
|
24
|
Kar R, Kellogg DL III and Roman LJ:
Oxidative stress induces phosphorylation of neuronal NOS in
cardiomyocytes through AMP-activated protein kinase (AMPK). Biochem
Biophys Res Commun. 459:393–397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kang S, Chemaly ER, Hajjar RJ and Lebeche
D: Resistin promotes cardiac hypertrophy via the AMP-activated
protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun
N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways.
J Biol Chem. 286:18465–18473. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Du JH, Xu N, Song Y, Xu M, Lu ZZ, Han C
and Zhang YY: AICAR stimulates IL-6 production via p38 MAPK in
cardiac fibroblasts in adult mice: A possible role for AMPK.
Biochem Biophys Res Commun. 337:1139–1144. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma Y, Wang J, Gao J, Yang H, Wang Y,
Manithody C, Li J and Rezaie AR: Antithrombin up-regulates
AMP-activated protein kinase signalling during myocardial
ischaemia/reperfusion injury. Thromb Haemost. 113:338–349. 2015.
View Article : Google Scholar
|
|
28
|
Li Y, Chen C, Yao F, Su Q, Liu D, Xue R,
Dai G, Fang R, Zeng J, Chen Y, et al: AMPK inhibits cardiac
hypertrophy by promoting autophagy via mTORC1. Arch Biochem
Biophys. 558:79–86. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang SY, Xu M, Ma XW, Xiao H and Zhang
YY: A distinct AMP-activated protein kinase phosphorylation site
characterizes cardiac hypertrophy induced by L-thyroxine and
angiotensin II. Clin Exp Pharmacol Physiol. 37:919–925. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Beauloye C, Bertrand L, Horman S and Hue
L: AMPK activation, a preventive therapeutic target in the
transition from cardiac injury to heart failure. Cardiovasc Res.
90:224–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamazaki KG, Gonzalez E and Zambon AC:
Crosstalk between the renin-angiotensin system and the advance
glycation end product axis in the heart: Role of the cardiac
fibroblast. J Cardiovasc Transl Res. 5:805–813. 2012. View Article : Google Scholar : PubMed/NCBI
|