Introduction
Dehydroepiandrosterone (DHEA), an intermediate
produced during the biosynthesis of steroids hormones, is secreted
by the adrenal cortex in an age-dependent manner (1). Previous studies have demonstrated
that DHEA exerts numerous beneficial effects, including prevention
of obesity (2), cancer (3), atherosclerosis (1) and age-induced changes to the brain
(4). The decrease in DHEA levels
with age is of significant clinical interest as DHEA reduction is
associated with physical health. In the United States, DHEA is
widely used as a non-prescribed dietary supplement (5).
The exact mechanisms of DHEA action remain unknown,
however, it may function through conversion to active steroid
hormones or effect certain metabolic enzymes, such as
glucose-6-phosphate dehydrogenase (G6PD) (6). DHEA is a pro-hormone and is rapidly
converted into testosterone and estradiol within peripheral target
tissues (7). A previous
investigation demonstrated that serum testosterone and estradiol
content were markedly increased in male rats following treatment
with DHEA (8). Additionally,
previous observations suggested that restoration of DHEA may alter
hormone contents resulting in anti-aging effects (9). However, the action of DHEA on the
biological characteristics of Leydig cells, which is major cell
type involved in DHEA biotransformation, remains unclear.
Previous studies have demonstrated that DHEA exerts
anti-proliferative effects in animal tumor models and in malignant
cell lines (10–12) through its inhibitory effects on
G6PD activity, which is essential for cell growth (6,11). A
previous study demonstrated that DHEA inhibits 3T3-L1 cell
proliferation, which was associated with G1 phase cell cycle arrest
(13). Zapata et al
(14) demonstrated that DHEA
inhibits mesodermal cell proliferation. In addition to metabolic
regulation, mitochondria are also critical for modulating other
cellular functions. Correa et al (15) demonstrated that DHEA inhibits
malate-glutamate oxidation by blocking Site I electron transport in
the respiratory chain, and induces mitochondrial swelling and
transmembrane electrical gradient collapse in isolated rat kidney
mitochondria. However, the mechanism of the effects of DHEA on
mitochondrial function is not fully understood.
It has been previously reported that the
biosynthesis and secretion of most androgen occurs in Leydig cells.
A previous study in Leydig cells suggested that functional changes
to the cells, rather than loss, cause the serum testosterone level
reduction (8). However, the
molecular mechanisms underlying the DHEA mode of action in primary
rat Leydig cells remain to be identified. The, the present study
aimed to investigate the effect of DHEA on cell proliferation and
mitochondrial function in primary rat Leydig cells. This
investigation is important to fully elucidate the cellular
mechanisms of DHEA activity and its effects in vivo.
Materials and methods
Animal and materials
Male Sprague-Dawley rats weighing 200±20 g were
purchased from Shanghai Experimental Animal Center of the Chinese
Academy of Sciences (Shanghai, China). DHEA, dimethyl sulfoxide
(DMSO), penicillin, streptomycin, trypsin and Percoll were
purchased from Sigma-Aldrich (St. Louis, MO, USA). Transferrin,
L-glutamine and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
(HEPES) were obtained from Amresco, LLC (Solon, OH, USA). Methyl
thiazolyl tetrazolium (MTT) was obtained from Sunshine Biotech
International Co., Ltd. (Bengbu, China). Dulbecco's modified
Eagle's medium (DMEM)-F12 and fetal bovine serum (FBS) were
purchased from Hyclone (GE Healthcare Life Sciences, Logan, UT,
USA). Click-iT EdU Imaging kits were purchased from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). TRIzol reagent was purchased
from Invitrogen (Thermo Fisher Scientific, Inc.). M-MLV reverse
transcriptase, RNase inhibitor and dNTP mixture were obtained from
Promega Corporation (Madison, WI, USA), and SYBR Green PCR Master
Mix was purchased from Takara Biotechnology Co., Ltd. (Dalian,
China). Propidium iodide cell cycle assay kit was obtained from
Multi Sciences (Lianke) Biotech Co., Ltd. (Hangzhou, China), and
5,5′, 6,6′-tetrachloro-1,1′, 3,3′-tetraethyl benzimidazol
carbocyanineiodide (JC-1) lipophiliccation kit, mitochondrial
membrane potential (ΔΨm) detection kits and bicinchoninic acid
protein assay kits were obtained from the Beyotime Institute of
Biotechnology (Haimen, China).
Primary Leydig cell culture
Male rats (2 months old) were housed individually
under a constant temperature of 25°C and 56–60% humidity, with a 12
h light-dark cycle. All animal handling procedures were performed
in strict accordance with guide for the Care and Use of Laboratory
Animals Central of the Nanjing Agricultural University (Nanjing,
China). The protocol was approved by the Institutional Animal Care
and Use Committee of the Nanjing Agricultural University (Nanjing,
China). Rats were killed by decapitation and Leydig cells were
isolated by enzymatic digestion and purified using discontinuous
Percoll gradient according to the method previously described by
Murugesan et al (16). The
purity of Leydig cells was assessed by 3β-hydroxysteroid
dehydrogenase histochemical localization according to the method
previously described by Aldred and Cooke (17), and using trypan blue dye exclusion
to determine the viability of purified Leydig cells. Subsequently,
cells were cultured in DMEM-F12 supplemented with 10% FBS, 5 mg/ml
transferrin, 2 mM L-glutamine, 1.75 mM HEPES, 100 IU/ml penicillin
and 100 mg/ml streptomycin in an atmosphere of 95% air and 5%
CO2 at 37°C.
Cell viability assay
Primary Leydig cells were seeded in 96-well plates
at a density of 1×104 cells/well and treated with 0.1,
1, 10, 50, 100 or 200 μM DHEA for 6, 12, 24 or 48 h before
MTT assay. In brief, 20 μl MTT (5 mg/ml) was added to each
well, and the plate was incubated for 4 h. Culture medium was
removed and the blue formazan crystals were dissolved in 50
μl DMSO, then the optical density (OD490 nm) was
measured using a model 550 microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA).
Morphological observation of cell
growth
Primary rat Leydig cells were plated in 6-well
plates at a density of 1×106 cells/well and incubated
for 24 h before treatment. The medium in each well was then
supplemented 0, 1, 50 or 100 μM DHEA. The cells were imaged
using a phase contrast microscope after 24 h.
EdU-based cell proliferation assays
Cell proliferation assays were performed using a
Click-iT EdU assay kit according to the manufacturer's
instructions. Briefly, the cells were plated at a density of
1×106 cells/well for 24 h. Fresh medium supplemented
with 0, 1, 50 or 100 μM DHEA was added in each well, the
cells were cultured for 24 h, then 100 μl
5′-ethynyl-2′-deoxyuridine (EdU) solution was added at a 50
μM final concentration for 2 h. Cells were harvested and
collected into 3 ml PBS containing 1% bovine serum albumin (GE
Healthcare Life Sciences), centrifuged at 1,500 × g for 10 min at
4°C and fixed with 100 μl 4% formaldehyde for 15 min.
Following formaldehyde fixation, cells were washed and then
incubated with 100 μl saponin-based permeabilization buffer
for 15 min. Cells were then incubated with 500 μl Click-iT
reaction buffer for 1 h and washed with 3 ml permeabilization
buffer. EdU-stained cells were mounted and imaged by fluorescence
microscopy.
Flow cytometry analysis of the cell
cycle
Primary rat Leydig cells were plated in 6-well
plates (1×106 cells/well) and treated with 0, 1, 50 or
100 μM DHEA for 6 h, 12 h, 24 h or 48 h. Cells were then
trypsinized, harvested and fixed in 1 ml 80% cold ethanol and
incubated at 4°C for 15 min. The cells were then centrifuged at
1500 × g for 5 min. The cell pellets were re-suspended in 500
μl propidium iodine (50 μg/ml) containing 5 U RNase
and incubated on ice for 15 min. Cell cycle distribution was
calculated from 10,000 cells with ModFit LT software (Verity
Software House, Inc., Topsham, ME, USA) using a FACSCaliber flow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Primary rat Leydig cells were cultured in 6-well
plates (1×106 cells/well) and treated with 1, 50 or 100
μM DHEA for 24 h. The cells were then harvested and total
RNA was extracted using TRIzol reagent according to the
manufacturer's instructions. RT was performed on 2 μg total
RNA by incubation for 1 h at 37°C in a 25 μl mixture
consisting of 100 U M-MLV reverse transcriptase, 8 U RNase
inhibitor, 0.5 μg of oligo dT, 50 mM Tris-HCl (pH 8.3), 3 mM
MgCl2, 75 mM KCl, 10 mM dithiothreitol and 0.8 mM
dNTP.
An aliquot of cDNA sample was mixed with 25
μl SYBR Green PCR Master Mix (Takara Biotechnology Co.,
Ltd.) and 10 pmol forward and reverse primers for β-actin (internal
control), cyclin-dependent kinase 2 (CDK2), cyclin A and cyclin B
(Table I), and then it was
subjected to PCR under standard conditions. All samples were
analyzed in duplicate using the ABI Prism 7300 Sequence Detection
System (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
programmed to conduct one cycle of 95°C for 1 min, then 40 cycles
of 95°C for 30 sec, 60°C for 30 sec and 72°C for 40 sec. The
2−ΔΔCq method was used to calculate the fold change in
mRNA levels (18). The primer
sequences were designed according to the published guidelines
(19) or using Primer Premier
(version 5; Premier Biosoft International, Palo Alto, CA, USA), and
synthesized by Takara Biotechnology Co., Ltd.
 | Table IPrimer sequence of β-actin and
targeted genes. |
Table I
Primer sequence of β-actin and
targeted genes.
| Gene | Genbank acession
number | Primer sequence
(5′-3′) | Orientation | Product size
(bp) |
|---|
| β-actin | NM_031144 |
CCCTGTGCTGCTCACCGA | Forward | 186 |
| |
ACAGTGTGGGTGACCCCGTC | Reverse | |
| Cyclin A | NM_053702 |
ATGTCAACCCCGAAAAAGTA | Forward | 154 |
| |
GGGACGTGCTCATCATCGTTTAT | Reverse | |
| Cyclin B | NM_171991 |
CTGACCCAAACCGCTGTA | Forward | 109 |
| |
GTCACTTCACGACCCTGT | Reverse | |
| CDK2 | NM_199501 |
CCCTTTCTTCCAGGATGTGA | Forward | 124 |
| |
AGCAGAAGGCTGACCTGTGT | Reverse | |
Quantification of mitochondria
The method used in the present study was modified
from the method described by Tang et al (20). Briefly, primary rat Leydig cells
were cultured in 6-well plates (1×106 cells/well) and
treated with 1 μM, 50 μM and 100 μM DHEA.
After 24 h, cells were fixed in 2.5% glutaraldehyde in 0.1 M sodium
phosphate (pH 7.4) and centrifuged at 3000 × g for 3 min at 4°C.
The cells were rinsed in 0.1 M sodium phosphate buffer (pH 7.4) and
post-fixed in 1% osmium tetroxide in Millonig's buffer. Then, cell
samples were processed by standard techniques for transmission
electron microscopy (TEM) (21).
Ultra-thin sections (60 nm) were stained with uranyl acetate and
lead citrate and visualized using an H-7650 transmission electron
microscope (Hitachi, Ltd., Tokyo, Japan). The number of
mitochondria were counted in 15 independent cells of 30 randomly
selected fields.
Mitochondrial membrane permeability
assay
A JC-1 ΔΨm detection kit was used to determine ΔΨm
according to the manufacturer's protocol. Briefly, 2×106
cells were collected and re-suspended in 0.5 ml medium. The cells
were mixed thoroughly with 0.5 ml JC-1 dye and incubated at 37°C
for 20 min in the dark prior to analysis using a flow cytometer (BD
Bioscience). The JC-1 monomer and polymer have an excitation
wavelength at 490 and 525 nm, and emission wavelength at 530 and
590 nm. Under low ΔΨm conditions, JC-1 predominantly exists as a
monomer and emits green fluorescence; while at high ΔΨm conditions,
JC-1 forms aggregates and emits a red fluorescence. The average
fluorescence intensity was calculated in 10 randomly selected
fields using Image Pro Plus software, version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA), and the 590/530 nm
fluorescence intensity ratio was used as an index for ΔΨm.
Analysis of succinate dehydrogenase
activity
Primary rat Leydig cells were cultured in 6-well
plates (1×106 cells/well) and treated with 1, 50 and 100
μM DHEA for 24 h. Following incubation, cells were harvested
and sonicated, then centrifuged at 2500 × g for 10 min at 4°C.
Supernatants were collected and succinate dehydrogenase activity
determined by a continuous spectrophotometric method, according to
manufacturer's instructions (Jianchen Biotechnology Institution,
Nanjing, China). Data were normalized to the sample protein
concentration, as determined by a protein assay kit.
Statistical analysis
Data were analyzed with one-way analysis of variance
and expressed as the mean ± standard error. Differences between
individual groups were analyzed by Duncan's multiple comparison
tests. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
with SPSS software for Windows (version 13; SPSS, Inc., Chicago,
IL, USA).
Results
Effect of DHEA on cell viability in
primary Leydig cells
The present study determined the effect of DHEA on
primary rat Leydig cell viability using the MTT method. As
presented in Table II, cell
viability was increased in the 50–200 μM DHEA-treated groups
at 6–48 h compared with the control group (P<0.01).
Additionally, cell viability was increased in presence of 0.1–10
μM DHEA at 24–48 h compared with the control group
(P<0.01).
| Table IIImpact of DHEA on primary rat Leydig
cell viability (optical density490). |
Table II
Impact of DHEA on primary rat Leydig
cell viability (optical density490).
| Group
(μmol/l) | Incubation time
|
|---|
| 6 h | 12 h | 24 h | 48 h |
|---|
| Control | 0.311±0.018 | 0.722±0.022 | 0.867±0.048 | 1.226±0.030 |
| 0.1 | 0.309±0.037 | 0.731±0.034 | 0.984±0.01b | 1.289±0.036b |
| 1.0 | 0.312±0.019 | 0.690±0.023 | 1.028±0.040b | 1.395±0.033b |
| 10.0 | 0.307±0.019 | 0.804±0.050 | 1.236±0.039b | 1.799±0.037b |
| 50.0 | 0.422±0.037b | 0.870±0.049b | 1.295±0.019b | 1.843±0.067b |
| 100.0 | 0.520±0.023b | 0.990±0.044b | 1.222±0.044b | 1.871±0.102b |
| 200.0 | 0.766±0.033b | 1.230±0.019b | 1.270±0.042b | 1.760±0.075b |
Effect of DHEA on cell proliferation in
primary Leydig cells
According to cell viability results, DHEA
concentrations of 1, 50 and 100 μM were used to treat
primary rat Leydig cells in all subsequent experiments. The results
demonstrated that DHEA significantly inhibited primary rat Leydig
cell growth (Fig. 1). To confirm
this result, a Click-iT EdU assay was used to examine the effect of
DHEA on cell proliferation. As demonstrated in Fig. 2, proliferating primary rat Leydig
cells that incorporated the nucleoside are red. Nuclei were
counterstained with Hoechst 33342 (blue) and pink color in the
merged image demonstrated the proliferating cells only. Our results
showed that DHEA treatment markedly inhibited primary rat Leydig
cell proliferation compared with controls in a dose-dependent
manner.
Effect of DHEA on the cell cycle in
primary Leydig cells
The cell cycle was evaluated by flow cytometry
(Fig. 3 and Table III). After 48 h exposure to 50
μM DHEA, the cell population in S phase was significantly
increased (P<0.01), whereas the population in G2/M was
significantly decreased (P<0.01) compared with the control
group. Furthermore, the S phase population was significantly
increased (P<0.01) and G2/M cell population decreased
(P<0.01) compared with the control group following 100 μM
DHEA incubation for 12–48 h in primary Leydig cells. No differences
in the cell cycle were detected in 1 μM DHEA-treated primary
Leydig cells compared with the control group (P>0.05).
| Table IIICell percentage following
DHEA-treatment. |
Table III
Cell percentage following
DHEA-treatment.
| Group |
Cell cycle
stage
|
|---|
6 h
| 12 h
| 24 h
| 48 h
|
|---|
| G0/G1 (%) | S (%) | G2/M (%) | G0/G1 (%) | S (%) | G2/M (%) | G0/G1 (%) | S (%) | G2/M (%) | G0/G1 (%) | S (%) | G2/M (%) |
|---|
| Vehicle | 56.61±1.89 | 27.61±0.37 | 15.78±2.265 | 35.98±1.38 | 38.55±1.78 | 25.46±0.48 | 58.96±0.96 | 23.30±0.70 | 17.75±0.26 | 69.16±2.36 | 21.55±2.03 | 9.29±0.77 |
| 1 µmol/l DHEA | 55.76±2.98 | 30.91±4.42 | 13.23±1.63 | 36.10±1.34 | 37.19±2.27 | 27.72±3.35 | 54.01±1.01 | 25.79±2.49 | 20.21±1.47 | 66.82±0.84 | 24.82±2.31 | 8.36±0.61 |
| 50 µmol/l DHEA | 57.62±1.23 | 30.41±1.45 | 11.97±0.24 | 35.18±1.57 | 40.29±1.51 | 24.54±0.15 | 58.10±2.04 | 27.88±1.62 | 14.03±1.24 | 68.29±0.72 | 26.98±0.53b | 4.73±0.66b |
| 100 µmol/l
DHEA | 56.20±1.58 | 31.66±1.39 | 11.99±0.47 | 36.71±1.84 | 48.34±0.99b | 14.95±1.72b | 55.60±3.86 | 38.86±4.02b | 5.55±1.03b | 67.12±0.50 | 31.10±1.11b | 1.78±0.29b |
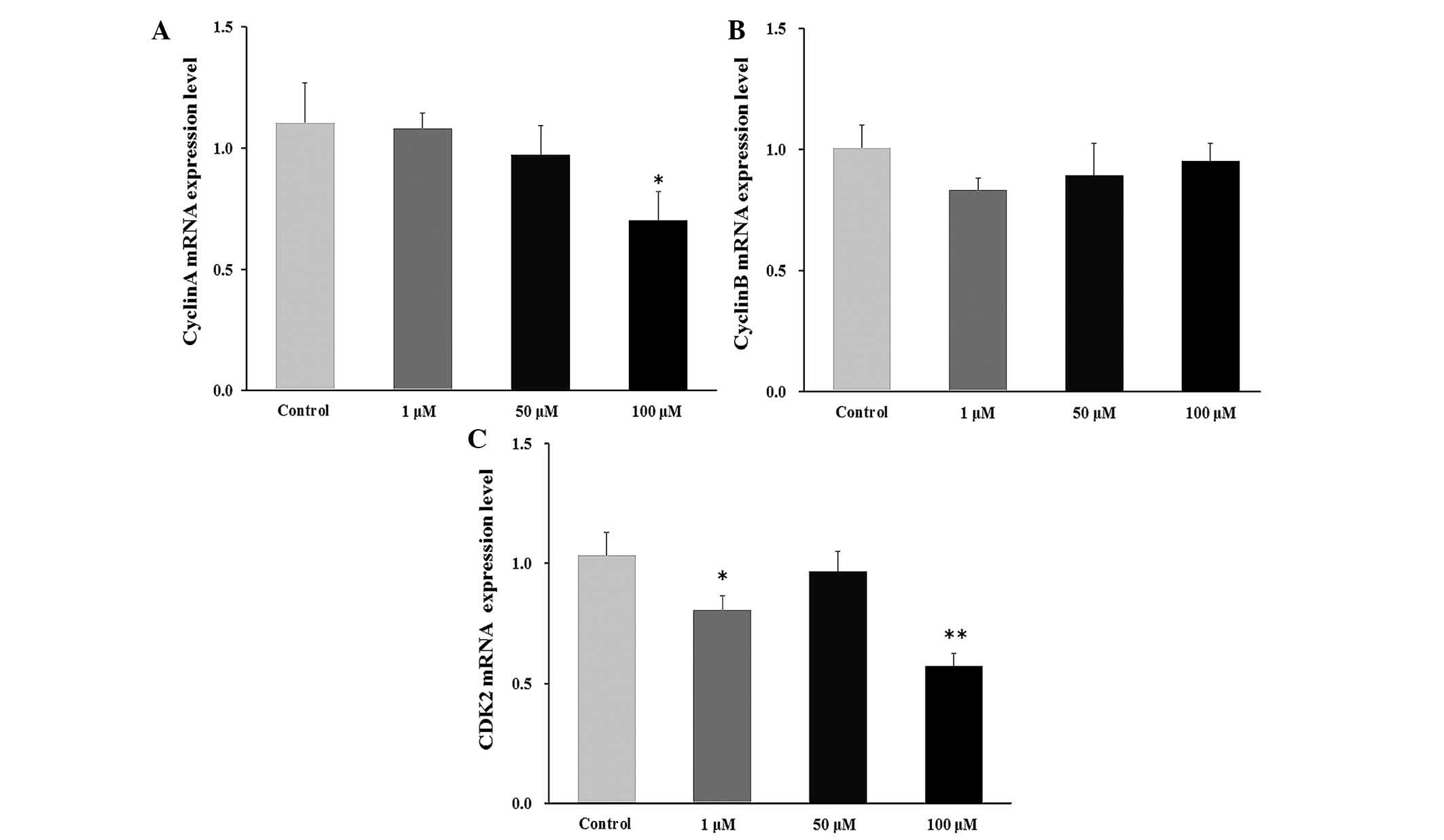
Effect of DHEA on cyclin mRNA levels in
primary Leydig cells
The level of cyclin mRNA was significantly decreases
in the 100 μM DHEA-treated group compared with the control
group (P<0.01; Fig. 4A). No
significant change was observed in the cyclin B mRNA level
(Fig. 4B), whereas the CDK2 mRNA
level was significantly decreased in the 1 μM (P<0.05)
and 100 μM (P<0.01) DHEA-treated groups compared with the
control group (Fig. 4C).
Morphological observations and
quantization of mitochondria
The histological organization of cultured primary
Leydig cells was not altered by DHEA treatment (Fig. 5). The number of mitochondria in 15
independent cells from 30 randomly selected fields were counted
with no significant change observed in the DHEA treated groups
compared with the control group (P<0.05; Fig. 5).
Effect of DHEA on the permeability of the
mitochondrial membrane in primary Leydig cells
ΔΨm was significantly decreased in 100 μM
DHEA-treated cells compared with control cells (P<0.01), whereas
only a marginal decrease was observed in 1 or 50 μM
DHEA-treated cells compared with control cells (P>0.05; Fig. 6). This result indicated that 100
μM DHEA treatment decreased the mitochondrial membrane
permeability in primary Leydig cells.
Effect of DHEA on succinate dehydrogenase
activity in primary Leydig cells
No significant difference in succinate dehydrogenase
activity was observed in the 1 μM DHEA-treated group
compared with the control group (P>0.05), whereas succinate
dehydrogenase activity was significantly increased in the 50 and
100 μM DHEA-treated groups compared with the control group
(P<0.01; Fig. 7).
Discussion
The major outcome of this study demonstrated that
DHEA inhibits cell proliferation through reducing cyclin mRNA
expression levels, whereas it markedly improves cell viability by
increasing the mitochondrial membrane permeability and succinate
dehydrogenase activity in primary Leydig cells. The majority of
endogenous DHEA is secreted by the adrenal cortex with some
produced by the testes and ovaries (1). DHEA possesses anti-proliferate
activity in various cell types (10–12).
However, little is known about its effect on primary rats Leydig
cell proliferation. In the present study, microscopy demonstrated
that DHEA treatment significantly inhibits primary Leydig cell
growth. Suzuki et al (22)
previously reported that DHEA modulates neuronal stem cell
proliferation, and Sicard et al (23) demonstrated that DHEA modulates
growth factor-induced proliferation in bovine adrenomedullary
tissue. The EdU assay is based on a copper-catalyzed covalent
reaction between a dye-conjugated azide and the alkyne group of EdU
(24–27), the product readily incorporates
into the DNA of replicating cells, including NIH 3T3 cells
(26,28) and mouse T-cells (29). The results of the current study
demonstrated that DHEA significantly decreases primary Leydig cell
proliferation in a dose-dependent manner, and this result is
consistent with the observations made using phase contrast
microscopy. It has been previously reported that DHEA inhibits the
proliferation of several types of cancer cells, including hepatoma,
prostate and cervical cancer (30–33).
A previous study also observed that DHEA induces proliferation of
estrogen and androgen receptor-positive breast cancer cells,
whereas it inhibits the proliferation of estrogen receptor-negative
cells (34). It is well recognized
that Leydig cells express estrogen and androgen receptors (35). However, López-Marure et al
(33) reported that DHEA decreases
both estrogen receptor-positive and -negative breast cancer cell
proliferation. Thus, based on the results of the current study, it
is speculated that the presence of estrogen or androgen receptors
may not be essential for the cell proliferation induced by DHEA,
and further study is required to precisely validate the effect of
DHEA on cell proliferation.
Certain evidence suggests that the inhibitory effect
of DHEA on cell proliferation is associated with the changes in the
phases of the cell cycle (31,33).
The present study demonstrated that 50 or 100 μM DHEA
treatment increased the S phase cell population and decreased the
G2/M population, indicating that DHEA inhibits primary rat Leydig
cell proliferation and causes cell cycle arrest in S phase. These
results are consistent with a previous study that suggested that
DHEA inhibits cell proliferation and blocks cell cycle progression
at the G1/S stage in 3T3-L1 cells (14). To investigate the effect of DHEA on
the cell cycle, the levels of cyclin mRNA were assessed. The
results demonstrated that cyclin A and CDK2 mRNA levels were
significantly decreased following 100 μM DHEA treatment,
whereas no significant change in cyclin B mRNA level was observed
in primary Leydig cells. In eukaryotes, the cell cycle is regulated
by cyclins, CDKs and CDK inhibitors. Particularly, cyclin A/CDK2
are involved in regulating the progression of S phase and cyclin
B/CDK1 are involved in regulating the progression of G2/M phase
(36). A previous study
demonstrated that the inhibitory effect of DHEA on the
proliferation of MCF-7 cells was associated with an arrest in G1
phase (33). Also, it has been
previously reported that the effect of DHEA on the MCF-7 cell cycle
is potentially dependent on DHEA metabolism to androstenediol
(37). Based on the results of the
current study, it is speculated that the inhibition of
proliferation induced by DHEA is associated with the decrease in
cyclin A and CDK2 mRNA levels, which in turn would then induce cell
cycle arrest in S phase, and this action of DHEA may occur with
conversion in primary Leydig cells. Further investigation of the
mRNA or protein levels of other factors, including CDK1 and cyclin
E, which are also associated with the cell cycle, is required to
validate this hypothesis more precisely.
It is notable that DHEA inhibited primary rat Leydig
cell proliferation, whereas it increased cell viability in a time-
and dose-dependent manner. In contrast with the results of the
present study, previous reports have demonstrated that DHEA
inhibits BV-2 cell viability (12,38),
however this effect was observed using ethanol as the solvent. It
has been previously reported that ethanol treatment results in a
slight decrease in cell viability. In the previous study, cell
viability was decreased by 8% at 1% ethanol, which was highest
final concentration used (13).
The present study demonstrated that the cell viability gradually
improved throughout the experimental period in the control group
(0.1% DMSO), which indicated that normal cell growth was
maintained. Thus, the use of different solvent may be the reason
for the discrepancy in the effect of DHEA on cell viability. To
further investigate the effect of DHEA on cell viability, the
mitochondria number, ΔΨm and succinate dehydrogenase activity were
subsequently analyzed. No significant change was detected in
mitochondria number and their configuration in primary Leydig cells
following DHEA treatment. However, previous studies have
demonstrated that DHEA affects mitochondrial number and
configuration in chicken hepatocytes (20) and the liver of male rats (39). DHEA also increases fatty acid
β-oxidation and enhances mitochondrial respiration, which may be
the cause of the decrease in mitochondria number in the rat or
chicken liver cells (20,39). The probable explanation for these
inconsistent results may be attributed to the different cell types
used in the studies.
Cell viability analysis was performed based on the
quantity of formazan produced from MTT by mitochondrial enzymes
(40). The double membrane
structure of the mitochondria blocks the entry of MTT into the
mitochondria. The result of the current study demonstrated that
DHEA treatment decreases the transmembrane ΔΨm of primary Leydig
cells, and the lowest ΔΨm was evident in the 100 μM
DHEA-treated group. This result indicated that the permeability of
the mitochondrial membrane was significantly increased in primary
Leydig cells treated with DHEA. Furthermore, this is consistent
with a previous study, in which the ΔΨm declined following
treatment of isolated kidney cortex mitochondria with high
concentrations of DHEA (15). Shen
et al (19) reported that
mitochondrial membrane permeability was significantly increased in
TM-3 cells following DHEA treatment. It has previously been
reported that DHEA administration to rats induces lipid
peroxidation in the liver and heart mitochondria (41). In this respect, it has previously
been demonstrated that mitochondrial membrane lipid peroxidation
causes an increase in permeability (42). These previous studies, at least
partially, indicate that increased primary Leydig cells viability
induced by DHEA is associated with the increased mitochondrial
membrane permeability. Succinate dehydrogenase is the only membrane
bound enzyme in the Krebs cycle, which directly links the oxidation
of succinate to the electron transport chain. The current study
demonstrated that succinate dehydrogenase activity was
significantly increased in primary Leydig cells following 50 or 100
μM DHEA treatment. DHEA has previously been demonstrated to
inhibit nicotinamide adenine dinucleotide-dependent mitochondrial
respiration and Complex I of the mitochondrial respiratory chain
(43), which results in ATP
depletion and the compensatory provision of ATP through glucose
oxidization (12). Sato et
al (44) reported that GLUT-4
protein expression level was increased and the glucose metabolic
signaling pathway was activated in skeletal muscle cells following
DHEA-treatment. Jahn et al (45) demonstrated that muscle glucose
oxidation was increased by 120% in diabetic Wistar rats treated
with DHEA. Thus, the effect of DHEA on succinate dehydrogenase
activity may be associated with its energy-wasting properties.
In summary, the results of the present study, at
least partially, demonstrate that DHEA-induced inhibition of
primary rat Leydig cell proliferation involves decreased cyclin
mRNA expression levels, which results in cell cycle arrest in S
phases. However, DHEA treatment improves cells viability by
modulating mitochondrial membrane permeability and succinate
dehydrogenase activity. However, further investigation is required
to fully elucidate the effect of DHEA on cell proliferation and
viability in primary rat Leydig cells.
Acknowledgments
The current work was supported by the Priority
Academic Program Development of Jiangsu Higher Education
Institutions.
References
|
1
|
Savineau JP, Mathan R and Dumas de la
Roque E: Role of DHEA in cardiovascular diseases. Biochem
Pharmacol. 85:718–726. 2013. View Article : Google Scholar
|
|
2
|
Sato K, Iemitsu M, Aizawa K, Mesaki N,
Ajisaka R and Fujita S: DHEA administration and exercise training
improves insulin resistance in obese rats. Nutr Metab (Lond).
9:472012. View Article : Google Scholar
|
|
3
|
Arnold JT, Gray NE, Jacobowitz K,
Viswanathan L, Cheung PW, McFann KK, Le H and Blackman MR: Human
prostate stromal cells stimulate increased PSA production in
DHEA-treated prostate cancer epithelial cells. J Steroid Biochem
Mol Biol. 111:240–246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kurita H, Maeshima H, Kida S, Matsuzaka H,
Shimano T, Nakano Y, Baba H, Suzuki T and Arai H: Serum
dehydroepiandrosterone (DHEA) and DHEA-sulfate (S) levels in
medicated patients with major depressive disorder compared with
controls. J Affect Disord. 146:205–212. 2013. View Article : Google Scholar
|
|
5
|
Legrain S and Girard L: Pharmacology and
therapeutic effects of dehydroepiandrosterone in older subjects.
Drugs Aging. 20:949–967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schwartz AG and Pashko LL:
Dehydroepiandrosterone, glucose-6-phosphate dehydrogenase and
longevity. Ageing Res Rev. 3:171–187. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Labrie F, Bélanger A, Bélanger P, Bérubé
R, Martel C, Cusan L, Gomez J, Candas B, Chaussade V, Castiel I, et
al: Metabolism of DHEA in postmenopausal women following
percutaneous administration. J Steroid Biochem Mol Biol.
103:178–188. 2007. View Article : Google Scholar
|
|
8
|
Song L, Tang X, Kong Y, Ma H and Zou S:
The expression of serum steroid sex hormones and steroidogenic
enzymes following intraperitoneal administration of
dehydroepiandrosterone (DHEA) in male rats. Steroids. 75:213–218.
2010. View Article : Google Scholar
|
|
9
|
Baulieu EE, Thomas G, Legrain S, Lahlou N,
Roger M, Debuire B, Faucounau V, Girard L, Hervy MP, Latour F, et
al: Dehydroepiandrosterone (DHEA), DHEA sulfate and aging:
Contribution of the DHEAge Study to a sociobiomedical issue. Proc
Natl Acad Sci USA. 97:4279–4284. 2000. View Article : Google Scholar
|
|
10
|
Dashtaki R, Whorton AR, Murphy TM, Chitano
P, Reed W and Kennedy TP: Dehydroepiandrosterone and analogs
inhibit DNA binding of AP-1 and airway smooth muscle proliferation.
J Pharmacol Exp Ther. 285:876–883. 1998.PubMed/NCBI
|
|
11
|
Di Monaco M, Pizzini A, Gatto V, Leonardi
L, Gallo M, Brignardello E and Boccuzzi G: Role of
glucose-6-phosphate dehydrogenase inhibition in the
antiproliferative effects of dehydroepiandrosterone on human breast
cancer cells. Brit J Cancer. 75:589–592. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang NC, Jeng KC, Ho WM and Hu ML: ATP
depletion is an important factor in DHEA-induced growth inhibition
and apoptosis in BV-2 cells. Life Sci. 70:1979–1988. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rice SP, Zhang L, Grennan-Jones F, Agarwal
N, Lewis MD, Rees DA and Ludgate M: Dehydroepiandrosterone (DHEA)
treatment in vitro inhibits adipogenesis in human omental but not
subcutaneous adipose tissue. Mol Cell Endocrinol. 320:51–57. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zapata E, Ventura JL, De la Cruz K,
Rodriguez E, Damián P, Massó F, Montaño LF and López-Marure R:
Dehydroepiandrosterone inhibits the proliferation of human
umbilical vein endothelial cells by enhancing the expression of p53
and p21, restricting the phosphorylation of retinoblastoma protein,
and is androgen-and estrogen-receptor independent. FEBS J.
272:1343–1353. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Correa F, Garcı́a N, Garcı́a G and Chávez
E: Dehydroepiandrosterone as an inducer of mitochondrial
permeability transition. J Steroid Biochem Mol Biol. 87:279–284.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murugesan P, Muthusamy T, Balasubramanian
K and Arunakaran J: Polychlorinated biphenyl (Aroclor 1254)
inhibits testosterone biosynthesis and antioxidant enzymes in
cultured rat Leydig cells. Reprod Toxicol. 25:447–454. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aldred LF and Cooke BA: The effect of cell
damage on the density and steroidogenic capacity of rat testis
Leydig cells, using an NADH exclusion test for determination of
viability. J Steroid Biochem. 18:411–414. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Shen X, Liu L, Yin F, Ma H and Zou S:
Effect of dehydroepiandrosterone on cell growth and mitochondrial
function in TM-3 cells. Gen Comp Endocrinol. 177:177–186. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang X, Ma H, Huang G, Miao J and Zou S:
The effect of dehydroepiandrosterone on lipogenic gene mRNA
expression in cultured primary chicken hepatocytes. Eur J Lipid Sci
Tech. 111:432–441. 2009. View Article : Google Scholar
|
|
21
|
Dickson GR: Practical Methods in Electron
Microscopy. Volume 3, part 1. Fixation, Dehydration and Embedding
of Biological Specimens. J Anat. 124(Part 2)1977.
|
|
22
|
Suzuki M, Wright LS, Marwah P, Lardy HA
and Svendsen CN: Mitotic and neurogenic effects of
dehydroepiandrosterone (DHEA) on human neural stem cell cultures
derived from the fetal cortex. Proc Natl Acad Sci USA.
101:3202–3207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sicard F, Ehrhart-Bornstein M, Corbeil D,
Sperber S, Krug AW, Ziegler CG, Rettori V, McCann SM and Bornstein
SR: Age-dependent regulation of chromaffin cell proliferation by
growth factors, dehydroepiandrosterone (DHEA), and DHEA sulfate.
Proc Natl Acad Sci USA. 104:2007–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Diermeier-Daucher S, Clarke ST, Hill D,
Vollmann-Zwerenz A, Bradford JA and Brockhoff G: Cell type specific
applicability of 5-ethynyl-2′-deoxyuridine (EdU) for dynamic
proliferation assessment in flow cytometry. Cytometry A.
75:535–546. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kotogány E, Dudits D, Horváth GV and
Ayaydin F: A rapid and robust assay for detection of S-phase cell
cycle progression in plant cells and tissues by using ethynyl
deoxyuridine. Plant Methods. 6:52010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Salic A and Mitchison TJ: A chemical
method for fast and sensitive detection of DNA synthesis in vivo.
Proc Natl Acad Sci USA. 105:2415–2420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Warren M, Puskarczyk K and Chapman SC:
Chick embryo proliferation studies using EdU labeling. Dev Dyn.
238:944–949. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chehrehasa F, Meedeniya AC, Dwyer P,
Abrahamsen G and Mackay-Sim A: EdU, a new thymidine analogue for
labelling proliferating cells in the nervous system. J Neurosci
Methods. 177:122–130. 2009. View Article : Google Scholar
|
|
29
|
Yu Y, Arora A, Min W, Roifman CM and
Grunebaum E: EdU incorporation is an alternative non-radioactive
assay to (3)H thymidine uptake for in vitro measurement of mice
T-cell proliferations. J Immunol Methods. 350:29–35. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arnold JT, Liu X, Allen JD, Le H, McFann
KK and Blackman MR: Androgen receptor or estrogen receptor-beta
blockade alters DHEA-, DHT- and E2 -induced
proliferation and PSA production in human prostate cancer cells.
Prostate. 67:1152–1162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Girón RA, Montaño LF, Escobar ML and
López-Marure R: Dehydroepiandrosterone inhibits the proliferation
and induces the death of HPV-positive and HPV-negative cervical
cancer cells through an androgen-and estrogen-receptor independent
mechanism. FEBS J. 276:5598–5609. 2009. View Article : Google Scholar
|
|
32
|
Ho HY, Cheng ML, Chiu HY, Weng SF and Chiu
DT: Dehydroepiandrosterone induces growth arrest of hepatoma cells
via alteration of mitochondrial gene expression and function. Int J
Oncol. 33:969–977. 2008.PubMed/NCBI
|
|
33
|
López-Marure R, Contreras PG and Dillon
JS: Effects of dehydroepiandrosterone on proliferation, migration,
and death of breast cancer cells. Eur J Pharmacol. 660:268–274.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toth-Fejel S, Cheek J, Calhoun K, Muller P
and Pommier RF: Estrogen and androgen receptors as comediators of
breast cancer cell proliferation: Providing a new therapeutic tool.
Arch Surg. 139:50–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kumar V, Balomajumder C and Roy P:
Disruption of LH-induced testosterone biosynthesis in testicular
Leydig cells by triclosan: Probable mechanism of action.
Toxicology. 250:124–131. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a
G2 arrest of the cell cycle and apoptosis accompanied with the
depletion of GSH. Cancer Lett. 270:40–55. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gebäck T, Schulz MMP, Koumoutsakos P and
Detmar M: TScratch: A novel and simple software tool for automated
analysis of monolayer wound healing assays. Biotechniques.
46:265–274. 2009.PubMed/NCBI
|
|
38
|
Yang NC, Jeng KC, Ho WM, Chou SJ and Hu
ML: DHEA inhibits cell growth and induces apoptosis in BV-2 cells
and the effects are inversely associated with glucose concentration
in the medium. J Steroid Biochem Mol Biol. 75:159–166. 2000.
View Article : Google Scholar
|
|
39
|
Bellei M, Battelli D, Fornieri C, Mori G,
Muscatello U, Lardy H and Bobyleva V: Changes in liver structure
and function after short-term and long-term treatment of rats with
dehydroepiandrosterone. J Nutr. 122:967–976. 1992.PubMed/NCBI
|
|
40
|
Bernas T and Dobrucki J: Mitochondrial and
nonmitochondrial reduction of MTT: Interaction of MTT with TMRE,
JC-1 and NAO mitochondrial fluorescent probes. Cytometry.
47:236–242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Swierczynski J and Mayer D:
Dehydroepiandrosterone-induced lipid peroxidation in rat liver
mitochondria. J Steroid Biochem Mol Biol. 58:599–603. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maciel EN, Vercesi AE and Castilho RF:
Oxidative stress in Ca (2+)-induced membrane permeability
transition in brain mitochondria. J Neurochem. 79:1237–1245. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Safiulina D, Peet N, Seppet E, Zharkovsky
A and Kaasik A: Dehydroepiandrosterone inhibits complex I of the
mitochondrial respiratory chain and is neurotoxic in vitro and in
vivo at high concentrations. Toxicol Sci. 93:348–356. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sato K, Iemitsu M, Aizawa K and Ajisaka R:
Testosterone and DHEA activate the glucose metabolism-related
signaling pathway in skeletal muscle. Am J Physiol Endocrinol
Metab. 294:E961–E968. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jahn MP, Jacob MH, Gomes LF, Duarte R,
Araújo AS, Belló-Klein A, Ribeiro MF and Kucharski LC: The effect
of long-term DHEA treatment on glucose metabolism, hydrogen
peroxide and thioredoxin levels in the skeletal muscle of diabetic
rats. J Steroid Biochem Mol Biol. 120:38–44. 2010. View Article : Google Scholar : PubMed/NCBI
|