Introduction
Multiple endocrine neoplasia type 2 (MEN2), also
known as Sipple's syndrome, is associated with medullary thyroid
cancer (MTC) and thyroid C cell hyperplasia. It is an autosomal
dominant genetic disease caused by a mutation in the RET
proto-oncogene (RET-PO) on chromosome 10 (1,2),
which results in 2 or more endocrine adenomas or hyperplasia in the
same patient, either concurrently or successively, and leads to the
clinical syndrome characterized by the hyperfunctioning of the
affected glands. Based on the clinical manifestation, MEN2 can be
divided into MEN2A, MEN2B and familial medullary thyroid carcinoma,
among which MEN2A is the most common subtype. MEN2A is
characterized by medullary thyroid carcinoma (MTC),
pheochromocytoma (PHEO) and hyperparathyroidism. Additionally, a
small number of patients exhibit skin lichen amyloidosis or
Hirschsprung's disease (3,4). MTC is often the first manifestation
of this subtype, with an incidence of close to 100% (5). When admitted, the majority patients
have already progressed to MTC or exhibit lymph node metastasis.
MTC is the predominant cause of death in patients with MEN2A
(6), and it has previously been
reported that 50% of patients are at risk of MTC recurrence
(7). However, the appearance of
MTC or MEN2A may differ among family members. Specifically, basic
lesions may be fully or not completely presented, the involved
endocrine glands may exhibit lesions that occur at different time
intervals (which may be as long as several years), and multiple
endocrine glands are occasionally involved and may exhibit
simultaneous onset. Currently, patients with MEN2A that exhibit MTC
as the early manifestation are often misdiagnosed. Therefore, the
present study analyzed the clinical data of one family with MEN2A
to investigate the clinical features and the significance of
RET-PO mutations.
Materials and methods
Clinical data
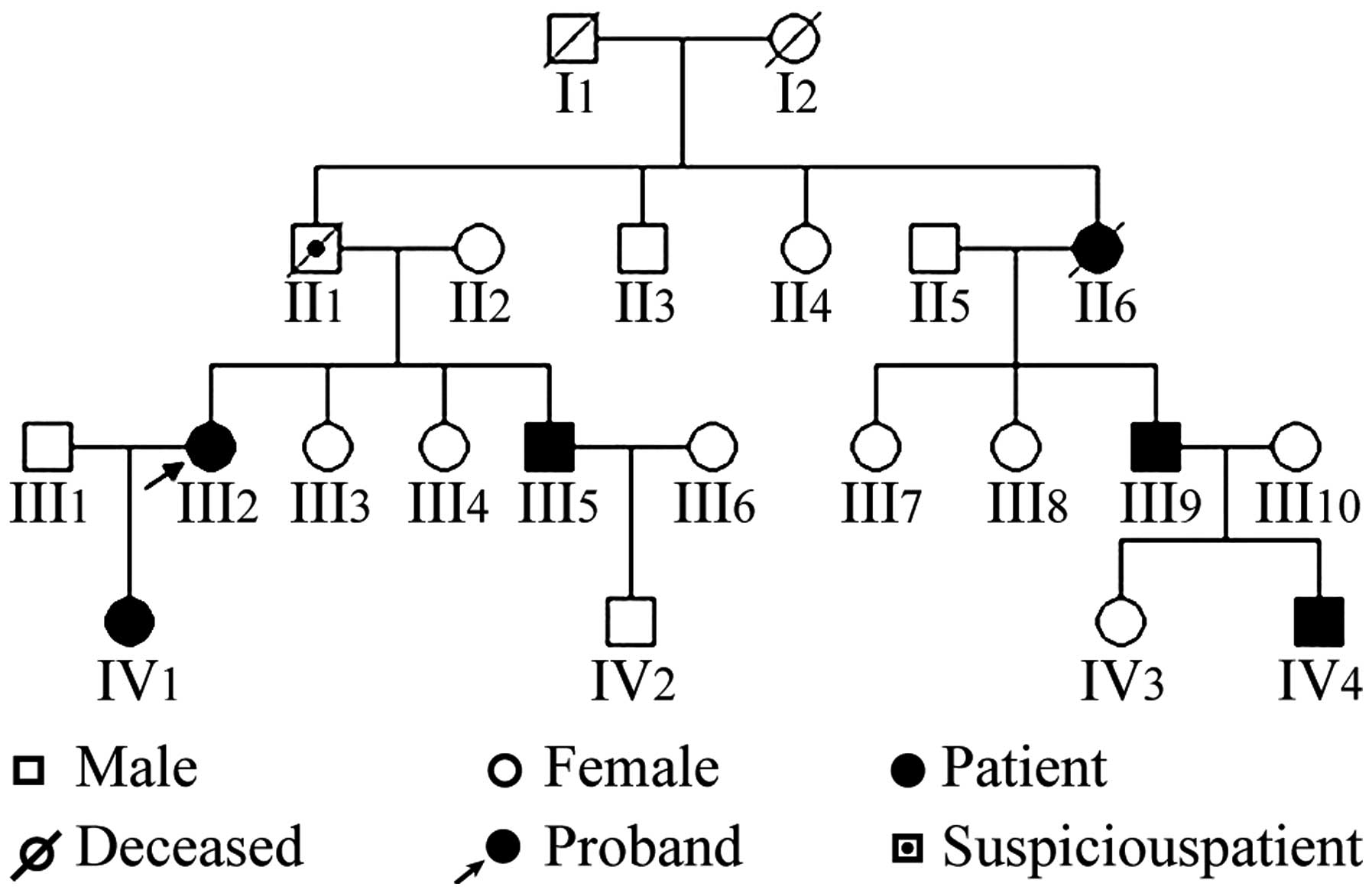
The family was a typical family with MEN2A, and the
family pedigree is presented in Fig.
1. The serum calcitonin (Ct), catecholamine (CA) and
carcinoembryonic antigen (CEA) levels of the patients (5 family
members: III2, III5, III9, IV1, and IV4) are presented in Table I.
 | Table ISerum Ct, CA and CEA of 5 members in
the multiple endocrine neoplasia type 2A pedigree family. |
Table I
Serum Ct, CA and CEA of 5 members in
the multiple endocrine neoplasia type 2A pedigree family.
| Patient | Gender | Preoperative
| Postoperative
|
|---|
| Ct (ng/l) | CA | Ct (ng/l) | CA | CEA (ng/ml) |
|---|
| III2 | Female | 637 | + | 175.00 | – | 6.59 |
| III5 | Male | Untested | Untested | <2.00 | – | 6.59 |
| III9 | Male | Untested | Untested | <2.00 | – | 1.17 |
| IV1 | Female | 56.20 | – | 4.10 | – | 3.75 |
| IV4 | Male | 169 | + | 14.10 | – | 1.36 |
Detection method for RET-PO
mutations
Peripheral blood (5 ml) with anticoagulant was
extracted from 13 family members, including the 5 patients (1
patient died). Briefly, all samples were anti coagulated with
sodium citrate. White blood cells were isolated from each blood
sample, and genomic DNA. Genomic DNA (200 ng) was amplified in a 50
µl reaction containing 10 mmol/l Tris-HCL (pH 8.4), 50
mmol/l KCL, 1.5 mmol/l MgCl2, 0.5 µmol/l each of
the specific primers, 100 µmol/l each of dATP, dGTP, dCTP
and dTTP, 2 units of Taq DNA polymerase (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The primers for exon 11 were synthesized
by Shenzhen Huada Gene Research Institute (Shenzhen, China). The
sequences were as follows: Forward, 5′-ACACCACCCCCACCCACAGAT-3′ and
reverse, 5′-AAGCTTGAAGGCATCCACGG-3′ (273 bp). The thermocycling
conditions were as follows: Denaturation at 94C for 5 min; 30
cycles of 94°C for 50 s, at 58–62°C for 50 sec, 72°C for 50 sec;
and a final extension at 72°C for 10 min. The DNA sequencing
analysis was conducted in Shenzhen Huada Gene Research
Institute.
Imaging and biochemical tests
Imaging tests, such as ultrasound/CT or emission
computed tomography, examined the thyroid, adrenal gland, and
parathyroid, at minimum. Serum Ct and CEA were analyzed using an
automated chemiluminescence immunoassay system (i2000; Abbott
Laboratories, Chicago, IL, USA) according to the manufacturer's
protocols. The serum CA was detected using the arsenazo III method
(8). The tribromoarsenazo was
provided by Sigma-Aldrich (St. Louis, MO, USA).
Follow-up
Follow-up was performed once every 3 months within 2
years following discharge the patient, then subsequently once every
6 months for 3 to 5 years, and once every 5 years thereafter. The
follow-up indicators measured included serum Ct, CEA, thyroid
function series, CA level and radiographic examination. Patients
with an RET mutation without PHEO were screened initially,
followed by examinations of CA levels and imaging annually
beginning at 20 years old. The present study was conducted in
accordance with the Declaration of Helsinki and with approval from
the Ethics Committee of Fudan University (Shanghai, China). Written
informed consent was obtained from all participants.
Results
Characteristics of the studied family
members
A total of 6 patients (4 males and 3 females) were
diagnosed with MEN2A and there was 1 suspected case. Among the 6
patients with MEN2A, 3 exhibited MTC and PHEO simultaneously.
Additionally, 1 patient died of hypertensive crisis during thyroid
surgery and was considered to have PHEO. The remain 2 patients
exhibited MTC only. The detailed clinical data are as follows.
The proband (III2) was a female and the onset age of
MEN2A was 28 years. The patient was admitted for 'episodic
headache, chest tightness and shortness of breath for 5 years' in
1991 and exhibited a blood pressure of 220/110 mmHg, serum Ct of
637 ng/l, and was positive for blood CA (+). The patient was
diagnosed as having PHEO in the right adrenal gland and a right
adrenal tumor resection was performed. A total thyroidectomy was
performed in 2001; the post-operative pathology demonstrated MTC
and the patient was finally diagnosed with MEN2A. Left adrenal PHEO
surgery was performed in 2008, and reviewed on 13 October 2014. The
serum Ct level was 175.00 ng/l (reference range: Male, <8.40
ng/l; female, <5.00 ng/l), CEA was 6.59 ng/ml (reference range,
0.00–5.00 ng/ml), and blood and urinary CA levels were normal.
B-ultrasonic examination of the thyroid (B-US) prompted resection
of the bilateral thyroids. The bilateral sides of the neck
exhibited lymphadenectasis, with calcification on the left side,
however no space-occupying lesion was present in the bilateral
adrenal glands. A computed tomography (CT) examination revealed no
lung, liver or bone metastasis, and the patient refused further
dissection toward the carotid lymph nodes.
The proband's father (II1) died of sudden high blood
pressure at 48 years old. According to Mendelian inheritance, MEN2A
was suspected. The proband's aunt (II6) underwent thyroid resection
at 51 years old and died of intraoperative hypertensive crisis. Her
pathological features suggested MTC. The proband's brother (III5)
underwent a physical examination at 28 years old that indicated
space-occupying masses in the bilateral adrenal glands and PHEO
surgery was performed in 2001. A B-US examination revealed a right
thyroid mass in 2003, and a right thyroid lobectomy and central
lymph node dissection was performed. Furthermore, pathological
analysis demonstrated MTC. An examination on 13 October 2014
revealed the following: Serum Ct, <2.00 ng/l; serum CEA, 6.59
ng/ml; and normal blood and urine CA levels. B-US prompted removal
of the right thyroid gland and the left thyroid gland exhibited no
lumps. No cervical lymph node metastasis was found in the bilateral
neck and no space-occupying lesion was found in the bilateral
adrenal glands. A CT examination did not detect any lung, liver or
bone metastasis.
The proband's cousin (III9) had a MEN2A onset age of
48 years old. He was admitted for 'left neck mass for 1 month' in
2009, and a left thyroid lobectomy and central lymph node
dissection was performed. The pathological analysis demonstrated
MTC. In 2010, a B-US review revealed a right thyroid mass, thus, a
right thyroid lobectomy and central lymph node dissection was
performed, and MTC was observed. An examination on 13 October 2014
revealed the following: Serum Ct, <2.00 ng/l; CEA, 1.17 ng/ml;
and normal blood and urine CA levels. B-US prompted removal of the
bilateral thyroids. No cervical lymph node metastasis was found in
the neck, and no space-occupying lesions were detected in the
bilateral adrenal glands. A CT examination did not detect any lung,
liver or bone metastasis.
The proband's daughter (IV1) had a MEN2A onset age
of 23 years old, and an RET C634Y mutation was detected via
genetic screening in July 2010. A B-US examination demonstrated
bilateral thyroid nodules and a CT examination observed a thickened
left adrenal gland with the possibility of hyperplasia. A
preoperative examination demonstrated the following: Serum Ct,
56.20 ng/l; and blood CA, negative. A total thyroidectomy was
performed on 14 September 2010 and MTC was detected. A review on 13
October 2014 demonstrated the following: Serum Ct, 4.10 ng/l; CEA,
3.75 ng/ml; and blood and urine CA levels, normal. B-US prompted a
resection of the bilateral thyroid and small lymph nodes were
observed in the neck, however no space-occupying lesion was
detected in the bilateral adrenal glands. A CT examination did not
detect any lung, liver or bone metastasis.
The proband's nephew (IV4) had an onset age of 19
years old and an RET C634Y mutation was identified by
genetic screening in July 2010. B-US revealed bilateral thyroid
nodules (multiple on the right side, including a right central
nodule K), posterior tubercles in the radix nasi of the
right thyroid gland, which may originate from the parathyroid, and
a right adrenal gland that exhibited a mixed tumor of the right
adrenal region with the possibility of pheochromocytoma plus
liquefaction. Preoperative serum Ct was 169.00 ng/l and blood CA
was positive. The patient underwent right adrenal PHEO surgery on
25 July 2010, and a total thyroidectomy on 4 August 2010, where MTC
was observed. A review on 13 October 2014 revealed the following:
Serum Ct, 14.10 ng/l; CEA, 1.36 ng/ml; and blood and urine CA
levels, normal. B-US prompted the removal of the bilateral thyroid.
No cervical lymph node metastasis was found in the neck and no
space-occupying lesion was observed in the bilateral adrenal
glands. A CT examination did not detect lung, liver or bone
metastasis.
Results of genetic tests
Among the 13 family members, 5 members (III2, III5,
III9, IV1, and IV4) exhibited the p.C634Y mutation. This was a
heterozygous missense mutation at locus 634 of exon 11 of the
RET gene resulting in a tyrosine to cysteine (TGC→TAC)
substitution in the protein amino acid sequence (Fig. 2). Of these cases, 2 were RET
mutation carriers (IV1 and IV4), but did not exhibit any clinical
symptoms. The remaining family members exhibited no RET
mutation, or clinical abnormalities based on imaging or Ct levels
indicating MTC. Additionally, 1 case (II6) was previously diagnosed
as MTC, but RET testing could not be performed due to death
of the patient. A pedigree analysis (Fig. 1) demonstrated that generations III
and IV were mutation carriers, and the transmission was consistent
with an inherited disease due to a dominant single gene.
Discussion
The RET-PO gene consists of 21 exons located
at chromosome 10q11.2 and encodes a tyrosine kinase receptor.
RET-PO mutations can lead to the conversion of a cysteine
residue to a different amino acid, which may lead to excessive cell
proliferation and potentially cancer development. Following the
observations in 1993 by Mulligan et al (9) and Donis-Keller et al (10) that an RET-PO mutation leads
to MEN2A, almost all MEN2A cases have been associated with an
RET-PO mutation (11,12).
A total of 93–98% of MEN2A cases exhibit a single-base
RET-PO mutation in exon 10 (codons 609, 611, 618 and 620) or
exon 11 (codon 634), with 87% exhibiting a mutation at codon 634.
The most common mutation type is C634R (TGC→CGC), accounting for
52%, followed by C634Y (TGC→TAC), accounting for 25% (13). In the current study, the family of
patients exhibited a C634Y mutation based on RET testing,
which is a common RET-PO mutation.
Numerous studies have confirmed that RET-PO
mutations are the predominant molecular etiology of MTC onset in
patients with MEN2A (9,14–16).
When RET mutations are confirmed, the probability of a
patient developing MTC is close to 100% (17). Thus, RET-PO detection is a
reliable method for diagnosing MEN2A, and is the gold standard for
early diagnosis (18). Its
accuracy is higher than that of Ct measurement and stimulation
tests. In 2009, it was recommended by the American Thyroid
Association (ATA) guidelines (3)
that all patients pathologically diagnosed with C cell hyperplasia,
MTC, MEN2A or autosomal dominant inheritance risk should undergo
RET-PO testing. Thus, in clinical practice, MTC patients
should be carefully examined and genetic testing should be
performed as early as possible. If germline RET-PO mutations
are detected in an individual without sporadic MTC, the patient is
likely to be a hereditary MTC proband.
MTC originates from thyroid parafollicular cells,
and unlike papillary thyroid carcinoma and follicular thyroid
carcinoma, the predominant, and only effective, MTC treatment is to
resect the primary tumor lesions and corresponding metastatic lymph
nodes as soon as possible (19).
There remains controversy regarding the optimal surgical methods
for thyroid primary tumors. Additionally, MTC has a low incidence
and diverse clinical manifestations, and factors that affect
prognosis vary between different studies (20). Some scholars propose that the most
appropriate method to treat MTC is total thyroidectomy and central
lymph node dissection, based on the theory that each parafollicular
cell of the RET-PO germline mutation carrier has the
potential for cancerization, and as such, all parafollicular cells
should be resected (21,22). Moreover, some scholars have
previously suggested that a patient with a unilateral lesion only
requires thyroid lobe and isthmus resection, and central lymph node
dissection on the affected side, and the contralateral gland should
be explored or resected (23). For
the patients with bilateral lesions, a total thyroidectomy should
be performed. The total thyroidectomy may result in increased
postoperative complications, including thyroid and parathyroid
insufficiency. For these reasons, the 2009 MTC treatment guidelines
established by the ATA (3), which
described the scope and extent of surgical resection toward typical
MTC patients, total thyroidectomy and preventive neck lymph node
dissection were recommended to reduce the recurrence of MTC.
Mutations in the RET gene are closely
associated with clinical manifestations and risk stratification of
MTC. Currently, the treatment of hereditary MTC is undergoing a
revolution from empirical treatment to individual treatment; the
latter emphasizes individualized treatment based on the codon
mutation site. In 2010, the North American Association of
Neuroendocrine Tumors published the MTC diagnosis and treatment
guidelines, which were divided into 3 groups based on the gene
mutation according to MTC invasion and high-risk characteristics
(24). RET-PO mutations
609, 768, 790, 791, 804 and 891 are included in the low-risk group,
which rarely develops into cancer before age 10, thus a preventive
thyroidectomy before 10 years of age is recommended. RET-PO
mutations 611, 618, 620 and 634 are included in the middle-risk
group, and a thyroidectomy is recommended before 5 years of age.
RET-PO mutations 883, 918 and 922 are included in the
high-risk group; patients in this group exhibit metastasis at 1
year of age, thus, thyroidectomy is recommended within 1 month of
reaching 6 months of age (25).
In the present study, 2 MEN2A gene mutation carriers
were identified by screening the 21 exons of RET-PO, and
preventive total thyroidectomy was performed. The postoperative
pathologies all confirmed MTC without lymph node metastasis. The
postoperative 4-year follow-up did not detect recurrence or
metastasis. This suggests that a preventive total thyroidectomy can
improve the cure rate before MTC is clinically observed, as lymph
node metastasis rarely occurs at this time, and it may also avoid
trauma caused by neck dissection. However, total thyroidectomy has
higher risks, for example, the incidences of complications,
including postoperative recurrent laryngeal nerve injury and
permanent hypoparathyroidism, are higher in children than in
adults. Accordingly, surgery in children should be performed by
experienced pediatric specialists to minimize complications.
PHEO in patients with MEN2A is associated with
bilateral adrenal tumors that may occur several years later, with
clinical manifestations including persistent or paroxysmal
hypertension, however, malignant tumors rarely occur or occur
outside of the adrenal glands. Approximately 30–50% of patients are
asymptomatic or experience sudden hypertension in thyroid
surgeries, and increased blood/urinary excretion of CA and its
metabolites may be the only abnormality in the early stage of this
disease (26,27). The diagnosis predominantly relies
on biochemical and imaging data, and 2 patients in the present
study exhibited elevated blood CA levels. If PHEO is present, it
should be resected with the application of an α-receptor blocker,
followed by the resection of other tumors to avoid induced
intraoperative hypertensive crisis and shock. Prior to surgery,
preparations including decompression, expansion, and heart rate
control should be conducted. Patients with bilateral adrenalectomy
should be given hormone replacement to prevent adrenal crisis. The
surgery should preserve a certain amount of normal adrenal tissues
to reduce the requirement of long-term hormone replacement therapy.
Additionally, radical thyroid resection should be performed
promptly.
As RET-PO mutations in MEN2A are strongly
correlated with clinical phenotypes of this disease, mutation
screening and analysis for MEN2A probands and family members may
suggest potential clinical phenotypes and enable a risk assessment,
thus, helping to determine the surgical timing of preventive
thyroidectomy, which may prevent or delay the progression of the
disease, and maximize clinical benefits. However, in China there
remain various difficulties associated with routine RET-PO
screening and the timing of preventive thyroidectomy cannot be
optimized in the short term. Previous investigations and the
results of the current study on a typical MEN2A family suggest that
family members with MEN2A should undergo RET-PO testing as
early as possible, asymptomatic carriers should be regularly
followed up, and surgery should be performed when lesions are
detected. This course of action is appropriate for use in
China.
References
|
1
|
Raue F and Frank-Raue K: Update multiple
endocrine neoplasia type 2. Fam Cancer. 9:449–557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wagner SM, Zhu S, Nicolescu AC and
Mulligan LM: Molecular mechanisms of RET receptor-mediated
oncogenesis in multiple endocrine neoplasia 2. Clinics (Sao Paulo).
67(Suppl 1): S77–S84. 2012. View Article : Google Scholar
|
|
3
|
American Thyroid Association Guidelines
Task Force; Kloos RT, Eng C, Evans DB, Francis GL, Gagel RF, Gharib
H, Moley JF, Pacini F, Ringel MD, et al: Medullary thyroid cancer:
Management guidelines of the American thyroid association. Thyroid.
19:565–612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qi XP, Ying RB, Ma JM, Liu WT, Du ZF, Fei
J, Yang CP, Song QZ, Jin HY, Chen ZG, et al: Case report: A p.C618S
RET proto-oncogene germline mutation in a large Chinese pedigree
with familial medullary thyroid carcinoma. Fam Cancer. 11:131–136.
2012. View Article : Google Scholar
|
|
5
|
Hibi Y, Ohye T, Ogawa K, Shimizu Y,
Shibata M, Kagawa C, Mizuno Y, Kurahashi H and Iwase K: A MEN2A
family with two asymptomatic carriers affected by unilateral renal
agenesis. Endocr J. 61:19–23. 2014. View Article : Google Scholar
|
|
6
|
Sakorafas GH, Friess H and Peros G: The
genetic basis of hereditary medullary thyroid cancer: Clinical
implications for the surgeon, with a particular emphasis on the
role of prophylactic thyroidectomy. Endocr Relat Cancer.
15:871–884. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roy M, Chen H and Sippel RS: Current
understanding and management of medullary thyroid cancer.
Oncologist. 18:1093–1100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferguson JW, Richard JJ, O'Laughlin JW and
Banks CV: Simultaneous spectrophotometric determination of calcium
and magnesium with chlorophosphonazo III. Anal Chem. 36:796–799.
1964. View Article : Google Scholar
|
|
9
|
Mulligan LM, Kwok JB, Healey CS, Elsdon
MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, et al:
Germ-line mutations of the RET proto-oncogene in multiple endocrine
neoplasia type 2A. Nature. 363:458–460. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Donis-Keller H, Dou S, Chi D, Toshima K,
Lairmore TC, Howe JR, Moley JF, Goodfellow P and Wells SA Jr:
Mutations in the RET proto-oncogene are associated with MEN 2A and
FMTC. Hum Mol Genet. 2:851–856. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gschwind A, Fischer OM and Ullrich A: The
discovery of receptor tyrosine kinases: Targets for cancer therapy.
Nat Rev Cancer. 4:361–370. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Machens A, Niccoli-Sire P, Hoegel J,
Frank-Raue K, van Vroonhoven TJ, Roeher HD, Wahl RA, Lamesch P,
Raue F, Conte-Devolx B, et al: Early malignant progression of
hereditary medullary thyroid cancer. N Engl J Med. 349:1517–1525.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pasini B, Ceccherini I and Romeo G: RET
mutations in human disease. Trends Genet. 12:138–144. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi XP, Chen XL, Ma JM, Du ZF, Fei J, Yang
CP, Cheng J, Song QZ, Han JS, Jin HY, et al: RET proto-oncogene
genetic screening of families with multiple endocrine neoplasia
type 2 optimizes diagnostic and clinical management in China.
Thyroid. 22:1257–1265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mulligan LM, Eng C, Healey CS, Clayton D,
Kwok JB, Gardner E, Ponder MA, Frilling A, Jackson CE, Lehnert H,
et al: Specific mutations of the RET proto-oncogene are related to
disease phenotype in MEN 2A and FMTC. Nat Genet. 6:70–74. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santoro M, Carlomagno F, Romano A, Bottaro
DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH,
et al: Activation of RET as a dominant transforming gene by
germline mutations of MEN2A and MEN2B. Science. 267:381–383. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sippel RS, Kunnimalaiyaan M and Chen H:
Current management of medullary thyroid cancer. Oncologist.
13:539–547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sawka AM, Jaeschke R, Singh RJ and Young
WF Jr: A comparison of biochemical tests for pheochromocytoma:
Measurement of fractionated plasma metanephrines compared with the
combination of 24-hour urinary metanephrines and catecholamines. J
Clin Endocrinol Metab. 88:553–558. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Milan SA, Sosa JA and Roman SA: Current
management of medullary thyroid cancer. Minerva Chir. 65:27–37.
2010.PubMed/NCBI
|
|
20
|
Roman S, Lin R and Sosa JA: Prognosis of
medullary thyroid carcinoma: Demographic, clinical and pathologic
predictors of survival in 1252 cases. Cancer. 107:2134–2142. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scollo C, Baudin E, Travagli JP, Caillou
B, Bellon N, Leboulleux S and Schlumberger M: Rationale for central
and bilateral lymph node dissection in sporadic and hereditary
medullary thyroid cancer. J Clin Endocrinol Metab. 88:2070–2075.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fleming JB, Lee JE, Bouvet M, Schultz PN,
Sherman SI, Sellin RV, Friend KE, Burgess MA, Cote GJ, Gagel RF and
Evans DB: Surgical strategy for the treatment of medullary thyroid
carcinoma. Ann Surg. 230:697–707. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
White ML, Gauger PG and Doherty GM:
Central lymph node dissection in differentiated thyroid cancer.
World J Surg. 31:895–904. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen H, Sippel RS, O'Dorisio MS, Vinik AI,
Lloyd RV and Pacak K; North American Neuroendocrine Tumor Society
(NANETS): The North American Neuroendocrine Tumor Society consensus
guideline for the diagnosis and management of neuroendocrine
tumors: Pheochromocytoma, paraganglioma, and medullary thyroid
cancer. Pancreas. 39:775–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Krampitz GW and Norton JA: RET gene
mutations (genotype and phenotype) of multiple endocrine neoplasia
type 2 and familial medullary thyroid carcinoma. Cancer.
120:1920–1931. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rodriguez JM, Balsalobre M, Ponce JL, Ríos
A, Torregrosa NM, Tebar J and Parrilla P: Pheochromocytoma in MEN
2A syndrome. Study of 54 patients. World J Surg. 32:2520–2526.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Inabnet WB, Caragliano P and Pertsemlidis
D: Pheochromocytoma: Inherited association, bilaterality and cortex
preservation. Surgery. 128:1007–1011. 2000. View Article : Google Scholar
|