Introduction
It is well known from epidemiological studies that
regular intake of aspirin reduces the risk of cancer of the
epithelial tissues, with the most profound protective effect being
observed against colon cancer (1–5).
Evidence that aspirin decreases the occurrence of epithelial cancer
is compelling; however, the pathways and proteins involved in this
process remain to be elucidated, and numerous mechanisms and
targets have been proposed (5–7).
Aspirin consists of two functional groups: The acetyl group and the
salicylate group, both of which have been implicated in its
anticancer effects. Early investigations in this area have
predominantly focused on the ability of aspirin to acetylate
cyclooxygenases (COX), leading to inactivation of enzyme activity,
as a primary mechanism to explain its anticancer effects (2). This is because inactivation of COX
induces decreased prostaglandin synthesis and inflammation, which
has been linked to decreased cancer occurrence. However, other
studies have reported that several COX-independent mechanisms
involving salicylic acid, the primary metabolite of aspirin, may
also contribute to its anticancer effects. Some of the direct
binding targets of salicylic acid that have been identified to date
include: IκB kinase β (8),
AMP-activated protein kinase (9),
high mobility group box 1 proteins (10) and cyclin dependent kinase 2
(11). It is argued that
modulation of the functional activity of these proteins by
salicylic acid may contribute to the anticancer effects of
aspirin.
Previous studies from our laboratory and others have
demonstrated that exposure of cancer cells to aspirin induces
acetylation of hundreds of proteins (12–15).
The acetylation targets of aspirin identified in our previous
studies included the tumor suppressor protein p53 (16,17),
enzymes of the glycolytic pathway, cytoskeletal proteins, histones,
and ribosomal and mitochondrial proteins (13). The enzymes that were identified in
the glycolytic pathway include aldolase, glyceraldehyde-3-phosphate
dehydrogenase, enolase, pyruvate kinase M2 and lactate
dehydrogenase A and B chains. In addition, we revealed that aspirin
acetylated glucose-6-phosphate dehydrogenase (G6PD) and
transketolase enzymes in the pentose phosphate pathway (13). Assays carried out for some of the
acetylated glycolytic pathway enzymes indicated that acetylation in
the majority of cases does not affect enzyme activity, thus
suggesting that it probably has a neutral impact on bulk protein
functions. However, acetylation of G6PD was associated with a
decrease in its enzyme activity, thus suggesting that aspirin
potentially modulates G6PD activity.
G6PD is an important regulatory enzyme in the
pentose phosphate pathway that produces ribose-5-phosphate, which
is essential for nucleic acid synthesis in rapidly dividing cells
(18,19). G6PD also produces the reducing
agent nicotinamide adenine dinucleotide phosphate (NADPH), which is
essential for the neutralization of oxygen free radicals and the
reductive biosynthesis of fatty acids. Therefore, G6PD has an
important role under normal physiological conditions, as well as in
cancer cell growth. Within human cells, two isoforms of G6PD have
been reported (20). G6PD isoform
a has a total of 545 amino acids, whereas isoform b
has a total of 515 amino acids. Isoform b is identical to
isoform a except that it lacks the first 30 amino acids in
the NH2 terminus. It has been reported that isoform b
represents the functionally active G6PD enzyme (20) and accordingly, the majority of
previous studies has reported on this isoform (21). Our earlier observation that aspirin
acetylated G6PD in HCT 116 colorectal cancer cells, leading to a
decrease in its enzyme activity, suggested the possibility that it
may have a role in the chemopreventive actions of aspirin (13). The present study extended these
earlier observations to HT-29 human colorectal cancer cells and
compared aspirin-mediated acet-ylation of G6PD and enzyme activity
between HCT 116 and HT-29 cells. The present study demonstrated
that compared with HCT 116 cells, HT-29 cells contained
significantly lower levels of acetylated G6PD; however, in both
cell types, G6PD protein expression levels were similar. To gain
insight into how aspirin decreases G6PD activity through
acetylation, mass spectrometry (MS) analysis was used to identify
the aspirin-acetylated sites on recombinant G6PD. The results
demonstrated that exposure of recombinant G6PD to aspirin induced
acetylation of 14 lysine residues. These lysine targets were found
to be localized throughout the G6PD protein, and included the
NAD-binding domain as well as the C-terminal domain. The important
targets of aspirin included lysine 235 (K235), which is present in
the highly conserved peptide RIDHYLGK (aa 228–235 in isoform
a) in G6PD. K235 in isoform a corresponds to K205 in
isoform b, which has previously been demonstrated to be
essential for catalysis (21). It
is likely that inhibition of G6PD enzyme activity by aspirin is due
to acetylation of this key lysine residue (K235 in isoform a
and K205 in isoform b). Lysine acetylation at K205 (or K235)
may prevent substrate binding (glucose-6-phosphate) into the active
site, rendering the enzyme catalytically inactive. Alternatively,
acetylation of other lysines in the nucleotide-binding domain may
also lead to decreased enzyme activity in cells treated with
aspirin.
Materials and methods
Materials
Cell culture reagents were purchased from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Aspirin was
obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-acetyl
lysine antibody (cat. no. 9441S; 1:5,000 dilution) was purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA), and
anti-G6PD antibody (cat. no. sc-46971; 1:2:000 dilution) was
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
The recombinant G6PD protein (isoform a) was obtained from
Origene Technologies, Inc. (Rockville, MD, USA). The G6PD activity
colorimetric assay kit was obtained from Biovision, Inc. (Milpitas,
CA, USA). All other chemicals were purchased from either
Sigma-Aldrich or Thermo Fisher Scientific, Inc.
Cell culture
HCT 116 and HT-29 colorectal cancer cells were
obtained from American Type Culture Collection (Manassas, VA, USA).
The cells were routinely cultured in McCoy's 5A medium (Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(Thermo Fisher Scientific, Inc.) at 37°C with 5% CO2
atmosphere. Cells were cultured for 12-16 h prior to the addition
of aspirin (0.25–2.5 mM) for the indicated times.
Preparation of cell lysates,
immunoprecipitation and western blotting
HCT 116 and HT-29 cells were grown for 12–16 h at
50% confluence, and treated with aspirin for the indicated times.
Cells were washed with phosphate-buffered saline and scraped in
lysis buffer, then protein concentrations were estimated by
Bradford protein assay, as previously described (13). A total of 200 µg total
protein was immunoprecipitated with agarose-conjugated anti-acetyl
lysine antibody overnight at 4°C, and was then washed three times
with lysis buffer. The agarose-bound proteins were eluted using
trimethylamine buffer and were immunoblotted with the anti-G6PD
antibody as previously described (13), and were immunoblotted with the
anti-G6PD antibody. Alternatively, 50 µg total proteins were
loaded onto an 8% SDS-polyacrylamide gel, and were immunoblotted
the anti-G6PD antibody. Subsequently, immunoreactive bands were
detected using a chemiluminescence system (Thermo Fisher
Scientific, Inc.).
G6PD assay
Subconfluent cells were left untreated or were
treated with the indicated concentrations of aspirin. The cells
were then lysed using the assay buffer. A total of 100 µg of
sample from each treatment condition was used for the G6PD assay,
which was performed in a 96-well plate according to the
manufacturer's protocol (Biovision, Inc.). Following termination of
the reaction, absorbance was measured at 450 nm.
Sample preparation for MS analysis, using
liquid chromatography-MS/MS
The recombinant G6PD isoform a (5 µg;
long form, NP_000393) was acetylated by incubating with 0.25 mM
aspirin for 12 h at room temperature, in the presence of 50 mM MOPS
(pH 7.4), in a final volume of 33 µl. For the negative
control, an equal amount of protein was left untreated under the
same conditions. An aliquot of acetylated protein was analyzed by
immunoblotting with anti-acetyl lysine antibody. The remaining
acetylated G6PD was subjected to MS as previous described (17).
Protein modeling
The FASTA sequence of recombinant G6PD (NP_000393)
was obtained from the National Center of Biotechnology Information
database. This sequence was then submitted to the SAM protein
modeling server (22). The
coordinates of the atoms obtained via email from the server were
transcribed and saved as a text file. The text file was then opened
in Rasmol protein modeling software (version 2.7.5). The locations
of lysine were mapped using command line in the software.
Statistical analysis
All experiments were repeated 3–6 times
independently. One-way analysis of variance followed by
Newman-Keuls multiple comparison tests were performed to compare
group differences to the control using Minitab software (version
16.0; Minitab, Inc., State College, PA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Correlation between G6PD acetylation
status and activity in HCT 116 and HT-29 cells
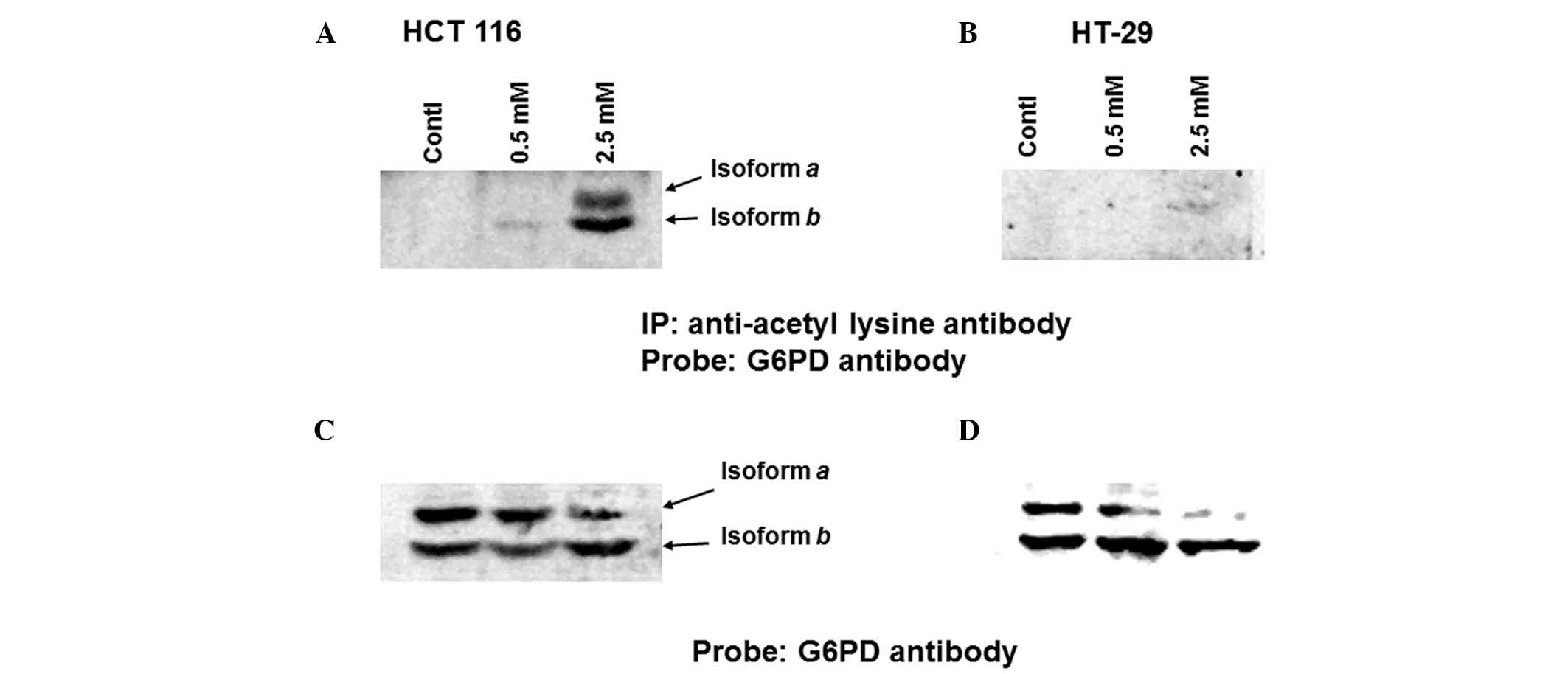
To compare the levels of aspirin-mediated G6PD
acetylation between HCT 116 and HT-29 cells, the cells were left
untreated or were treated with two different concentrations of
aspirin (0.5 and 2.5 mM) for 24 h. The lysates were then
immunoprecipitated with anti-acetyl lysine antibody, and
immunoblotted with anti-G6PD antibody. As shown in Fig. 1A, aspirin-induced acetylation of
G6PD was detected at 0.5 mM in HCT 116 cells; however, markedly
increased levels were detected at 2.5 mM. Conversely, in the HT-29
cells, G6PD acetylation was not observed at all at 0.5 mM, and was
barely detected at 2.5 mM (Fig.
1B). As presented in Fig. 1C and
D both isoforms of G6PD (isoforms a and b) were
detected in HCT 116 and HT-29 cells. G6PD activity was then
detected in lysates prepared from both HCT 116 and HT-29 cells.
Aspirin progressively inhibited G6PD activity in HCT 116 cells
beginning at 0.5 mM; however, inhibition was not so strong in HT-29
cells (Fig. 2). These results
suggest that aspirin-mediated acetylation may lead to inhibition of
G6PD activity within the cellular milieu.
In vitro acetylation of recombinant G6PD
protein by aspirin
The human G6PD enzyme (isoform a, long form)
has a total of 29 lysine residue in its 545 amino acid long
sequence. To determine which of the lysines are targeted by
aspirin, the commercially obtained purified recombinant G6PD was
acetylated with aspirin in vitro, and the product was
subjected to MS analysis. Briefly, 5 µg purified G6PD
protein was incubated with 0.25 mM aspirin for 12 h; an aliquot (5
ng) of the sample was initially analyzed by immunoblotting with
anti-acetyl lysine antibody to ensure acetylation. As shown in
Fig. 3A, anti-acetyl antibodies
prominently detected a 56 kDa protein in the aspirin-treated
conditions, which was not detected in the untreated control
group.
Identification of aspirin-acetylated
sites on recombinant G6PD
An aliquot of the in vitro acetylated G6PD
(isoform a), as presented in the blot in Fig. 3A, was subjected to MS analysis for
the identification of acetylation sites. Based on the MS analysis,
a total of 14 lysine residues (K77, K112, K119, K201, K235, K390,
K396, K416, K438, K459, K462, K527, K538, K544) were identified as
targets of aspirin-mediated acetylation. The spectra obtained for
three of the acetylated lysine-containing peptides are shown in
Fig. 3B–D. The spectra for other
acetylated peptides are not shown. The acetylated peptides
following digestion with trypsin and chymotrypsin are shown in
Table I. The location of
aspirin-acetylated lysine residues identified in the present study,
and the comparison to naturally acetylated lysines on G6PD are
shown in Table II. The peptide
digests covered 86% of the total G6PD sequence. Notably, the lysine
at position 235 (K235) (corresponding to K205 in G6PD isoform
b), which is located in the active site of the enzyme within
the evolutionarily conserved peptide RIDHYLGK (aa 228–235), is
acetylated by aspirin. K205 (in isoform b) has previously
been demonstrated to be important for catalysis (21). It is likely that acetylation of
this residue is responsible for the observed decrease in G6PD
activity in HCT 116 cells.
 | Table IAspirin-acetylated peptides
identified from glucose-6-phosphate dehydrogenase after trypsin and
chymotrypsin digestion. |
Table I
Aspirin-acetylated peptides
identified from glucose-6-phosphate dehydrogenase after trypsin and
chymotrypsin digestion.
| Acetylated
position | Score | Protease | Peptide |
|---|
| 77 | 43 | Trypsin | K*IYPTIWWLFR |
| 33 | Chymo | AKKK*IYPTIW |
| 112 | 31 | Trypsin | K*QSEPFFK |
| 25 | Chymo | TVADIRK*QSEPF |
| 119 | 37 | Trypsin | KQSEPFFK* |
| 31 | Chymo | FK*ATPEEKL31 |
| 31 | Chymo | K*ATPEEKL |
| 201 | 56 | Trypsin | IIVEK*PFGR |
| 26 | Chymo | NRIIVEK*PF |
| 235 | 33 | Trypsin | IDHYLGK* |
| 26 | Chymo | LGK*EMVQNL |
| 390 | 46 | Trypsin | CGK*ALNER |
| 396 | 26 | Chymo | NERK*AEVRLQF |
| 416 | 24 | Trypsin |
LQFHDVAGDIFHQQCK*R |
| 27 | Chymo | HQQCK*RNEL |
| 438 | 47 | Trypsin |
K*PGMFFNPEESELDLTYGNR |
| 459 | 28 | Chymo | K*NVKLPDAY |
| 43 | Chymo | K*NVKLPDAYERL |
| 462 | 22 | Trypsin | NVK*LPDAYER |
| 527 | 78 | Trypsin | GPTEADELMK*R |
| 61 | Trypsin | GPTEADELMK*R |
| 538 | 50 | Trypsin |
VGFQYEGTYK*WVNPHK |
| 544 | 26 | Chymo | KWVNPHK*L |
| Table IILocation of aspirin-acetylated lysine
residues identified in the present study and the naturally
acetylated lysines on G6PD (25). |
Table II
Location of aspirin-acetylated lysine
residues identified in the present study and the naturally
acetylated lysines on G6PD (25).
| Aspirin-acetylated
lysine sites in isoform a (long) | Corresponding
lysine sites in isoform b (short) | Naturally
acetylated sites |
|---|
| K77 | K47 | |
| K112 | K82 | |
| K119 | K89 | K89 |
| K201 | K171 | K171 |
| K235 | K205a | |
| K390 | K360 | |
| K396 | K366 | |
| K416 | K386 | K386 |
| | K403b |
| K438 | K408 | |
| K459 | K429 | |
| K462 | K432 | K432 |
| K527 | K497 | K497 |
| K538 | K508 | |
| K544 | K514 | K514 |
Molecular modeling of recombinant G6PD (NCBI
accession no: NP_000393) demonstrated that the majority of the
aspirin-acetylated lysine residues are surface-exposed/solvent
accessible (Fig. 4). The ability
of aspirin to acetylate G6PD in vitro suggests that it is a
non-enzymatic chemical reaction, consistent with its ability to
acetylate numerous other proteins in-vitro (23).
 | Figure 43-Dimension space-filling model of
recombinant glucose-6-phosphate dehydrogenase (G6PD; NP_000393), is
shown. The location of aspirin-acetylated lysine residues are
highlighted in blue (K77, K112, K119, K201, K235, K390, K396, K416,
K438, K459, K462, K527, K538, K544). |
Discussion
The housekeeping enzyme G6PD is the major regulatory
enzyme in the pentose phosphate pathway, which catalyzes the
conversion of glucose-6-phosphate to 6-phosphoglucono-δ-lactone
with a concomitant reduction of NADP+. NADPH thus
generated is essential for the neutralization of oxidative stress
within the cellular milieu. In addition, the pentose phosphate
pathway generates ribose sugars, which are required for nucleic
acid synthesis, and has a major role in rapidly dividing normal
cells, as well as in cancer cell growth. Our previous study
demonstrated that aspirin acetylates G6PD in HCT 116 cells, and
this was associated with a decrease in its enzyme activity;
however, the mechanism underlying this inhibition was not clearly
identified (13). The aim of the
present study was two-fold: i) To compare aspirin-mediated G6PD
acetylation and enzyme activity between HCT 116 and HT-29 cells;
and ii) identify the aspirin-mediated acetylation targets in
recombinant G6PD. The results demonstrated that exposure of HCT 116
cells to aspirin induced greater acetylation of G6PD compared with
in HT-29 cells; accordingly, increased inhibition of G6PD activity
was also observed in the HCT 116 cells. Although acetylated G6PD
levels were lower in HT-29 cells, the G6PD protein levels (both
isoform a and b) were similar in both HCT 116 and
HT-29 cells. The decreased levels of G6PD acetylation in HT-29
cells may be associated with a lower aspirin uptake, or more rapid
hydrolysis of aspirin within the cells due to the action of
esterases. Through MS analysis of in vitro acetylated
recombinant G6PD (isoform a), a total of 14 lysine residues
(K77, K112, K119, K201, K235, K390, K396, K416, K438, K459, K462,
K527, K538, K544) were identified as targets of aspirin-mediated
acetylation. These sites are dispersed throughout the length of the
G6PD protein and are located in both the NAD-binding (aa 65-240)
and the C-terminal domains (aa 242-536) (24). These results suggested that, in
some cancer cell lines, aspirin may cause a direct inhibition of
G6PD activity by modifying the protein via acetylation; and this
may potentially contribute to reduced synthesis of ribose sugars
and NADPH in cancer cells.
Within cells, G6PD exists as a mixture of monomer,
dimer, tetramer and hexamer; but only the dimeric and tetrameric
forms of the enzyme are catalytically active (25). The active enzyme exists in a
dimer↔tetramer equilibrium. It has previously been reported that
G6PD isoform b (short form) is naturally acetylated at seven
lysine residues: K89, K171, K386, K403, K432, K497 and K514
(25,26). These modified lysines in isoform
b correspond to K119, K201, K416, K433, K462, K527 and K544
in G6PD isoform a (long form). Among the naturally
acetylated sites, and aspirin-acetylated sites, six of the seven
sites appear to be common: K119, K201, K416, K462, K527 and K544.
Notably, the acetylation of K235 by aspirin in G6PD isoform
a was observed in the present study. This corresponds to
K205 in isoform b; this lysine residue has been demonstrated
to be part of the active site of the enzyme, and is important in
catalysis (21). K205 is located
within the highly conserved peptide RIDHYLGK (residues 198–205,
single letter amino acid code in isoform b) (27). Notably, in a previous study, it was
reported that exposure of purified yeast G6PD to aspirin induced
acetylation of a lysine within the G6PD peptide sequence IDHYLGK
(aa 185–191), which resulted in inactivation of the enzyme activity
(28). These previous reports
(21,28) along with the present findings,
suggested that acetylation of this critical lysine (K205 in isoform
b, and K235 in isoform a) important for catalysis may
affect substrate binding (glucose-6-phosphate) and contribute to
the aspirin-mediated inhibition of G6PD activity observed in HCT
116 cells. Alternatively, acetylation of other lysines may affect
the affinity of NADP binding to G6PD, or simply may affect the
dimer/tetramer formation required for keeping the enzyme in the
active configuration. Molecular modeling of G6PD isoform a
suggested that the majority of aspirin-acetylated lysines are
surface-exposed/solvent accessible (Fig. 4). Similar molecular modeling of
isoform b suggested that the corresponding amino acids
(including K205 in the active site) are also surface-exposed (data
not shown).
Wang et al (25) revealed that acetylation of K403 (in
G6PD isoform b), mediated by cellular lysine
acetyltransferases, negatively regulated enzyme activity. In
addition, it was demonstrated that K403-acetylated G6PD is
incapable of forming active dimers and displays a complete loss of
activity. The K403 lysine in isoform b corresponds to K433
in isoform a, and in the present MS analysis, this lysine
was not identified as a target of aspirin. However, other lysine
residues in close proximity, including K416 and K438, were
acetylated following treatment with aspirin.
The present study demonstrated that aspirin
acetylates G6PD, and this is associated with decreased enzyme
activity, potentially due to modification of a lysine within the
highly conserved peptide RIDHYLGK. These are important findings in
the context of the known role of G6PD in cancer cell growth. G6PD
has been reported to be overexpressed in numerous types of cancer,
including breast, colon, endometrial, cervical, prostate and lung
cancers (18,19,29–32).
Both NADPH and ribose-5-phosphates are essential products generated
in the pentose phosphate pathway, which are important for cancer
cell growth, through their contribution to nucleic acid
biosynthesis and protection against oxidative stress. In addition,
it has been reported that ectopic expression of G6PD in NIH3T3
cells significantly increased intracellular levels of NADPH and
promoted anchorage-independent cell growth (33,34).
Due to its important role in cancer development, and increased
expression in cancer, it is argued that G6PD may be a potential
therapeutic target for cancer treatment (19). Based on the findings of the present
study in HCT 116 cells, and the acetylation of K235 in isoform
a G6PD (equivalent to K205 in isoform b), it is
likely that aspirin may inhibit G6PD activity in other cancer cell
types; and therefore, may contribute to decreased cancer cell
growth through reduced synthesis of ribose sugars and NADPH.
Acknowledgments
Support from the Translational Cancer Research Seed
Grant, funded as the 2010 Research Initiative Center by the State
of South Dakota, and from NIH (grant no. 5RO3CA133061-02) to GJB is
gratefully acknowledged. The authors would also like to thank Mr.
Raghavender Chivukula (Texas Tech University Health Science Center)
for helpful discussions, and Dr. Fred Hagen (University of
Rochester Medical Center, Rochester, NY) for carrying out the MS
analysis.
References
|
1
|
Kaiser J: Will an aspirin a day keep
cancer away? Science. 337:1471–1473. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thun MJ, Jacobs EJ and Patrono C: The role
of aspirin in cancer prevention. Nat Rev Clin Oncol. 9:259–267.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chan AT, Arber N, Burn J, Chia WK, Elwood
P, Hull MA, Logan RF, Rothwell PM, Schrör K and Baron JA: Aspirin
in the chemoprevention of colorectal neoplasia: An overview. Cancer
Prev Res (Phila). 5:164–178. 2012. View Article : Google Scholar
|
|
4
|
Rothwell PM, Price JF, Fowkes FG,
Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M,
Mehta Z and Meade TW: Short-term effects of daily aspirin on cancer
incidence, mortality, and non-vascular death: Analysis of the time
course of risks and benefits in 51 randomised controlled trials.
Lancet. 379:1602–1612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferrández A, Piazuelo E and Castells A:
Aspirin and the prevention of colorectal cancer. Best Pract Res
Clin Gastroenterol. 26:185–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alfonso L, Ai G, Spitale RC and Bhat GJ:
Molecular targets of aspirin and cancer prevention. Br J Cancer.
111:61–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dovizio M, Bruno A, Tacconelli S and
Patrignani P: Mode of action of aspirin as a chemopreventive agent.
Recent Results Cancer Res. 191:39–65. 2013. View Article : Google Scholar
|
|
8
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hawley SA, Fullerton MD, Ross FA,
Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green
KA, Mustard KJ, et al: The ancient drug salicylate directly
activates AMP-activated protein kinase. Science. 336:918–922. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi HW, Tian M, Song F, Venereau E, Preti
A, Park SW, Hamilton K, Swapna GV, Manohar M, Moreau M, et al:
Aspirin's active metabolite salicylic acid targets high mobility
group Box 1 to modulate inflammatory responses. Mol Med.
21:526–535. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dachineni R, Ai G, Kumar DR, Sadhu SS,
Tummala H and Bhat GJ: Cyclin A2 and CDK2 as novel targets of
aspirin and salicylic acid: A potential role in cancer prevention.
Mol Cancer Res. 14:241–252. 2016. View Article : Google Scholar
|
|
12
|
Alfonso LF, Srivenugopal KS and Bhat GJ:
Does aspirin acetylate multiple cellular proteins? (Review). Mol
Med Rep. 2:533–537. 2009.PubMed/NCBI
|
|
13
|
Marimuthu S, Chivukula RS, Alfonso LF,
Moridani M, Hagen FK and Bhat GJ: Aspirin acetylates multiple
cellular proteins in HCT-116 colon cancer cells: Identification of
novel targets. Int J Oncol. 39:1273–1283. 2011.PubMed/NCBI
|
|
14
|
Bateman LA, Zaro BW, Miller SM and Pratt
MR: An alkyne-aspirin chemical reporter for the detection of
aspirin-dependent protein modification in living cells. J Am Chem
Soc. 135:14568–14573. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Zhang CJ, Zhang J, He Y, Lee YM,
Chen S, Lim TK, Ng S, Shen HM and Lin Q: Mapping sites of
aspirin-induced acetylations in live cells by quantitative
acid-cleavable activity-based protein profiling (QA-ABPP). Sci Rep.
5:78962015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alfonso LF, Srivenugopal KS, Arumugam TV,
Abbruscato TJ, Weidanz JA and Bhat GJ: Aspirin inhibits
camptothecin-induced p21CIP1 levels and potentiates apoptosis in
human breast cancer cells. Int J Oncol. 34:597–608. 2009.PubMed/NCBI
|
|
17
|
Ai G, Dachineni R, Kumar DR, Marimuthu S,
Alfonso LF and Bhat GJ: Aspirin acetylates wild type and mutant p53
in colon cancer cells: Identification of aspirin acetylated sites
on recombinant p53. Tumor Biol. Nov 23–2015.Epub ahead of
print.
|
|
18
|
Stanton RC: Glucose-6-phosphate
dehydrogenase, NADPH, and cell survival. IUBMB Life. 64:362–369.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Furuta E, Okuda H, Kobayashi A and Watabe
K: Metabolic genes in cancer: Their roles in tumor progression and
clinical implications. Biochim Biophys Acta. 1805:141–152.
2010.PubMed/NCBI
|
|
20
|
Kanno H, Kondoh T and Yoshida A: 5′
Structure and expression of human glucose-6-phosphate dehydrogenase
mRNA. DNA Cell Biol. 12:209–215. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bautista JM, Mason PJ and Luzzatto L:
Human glucose-6-phosphate dehydrogenase. Lysine 205 is dispensable
for substrate binding but essential for catalysis. FEBS Lett.
366:61–64. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karplus K: SAM-T08, HMM-based protein
structure prediction. Nucleic Acids Res. 37:W492–W497. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pinckard RN, Hawkins D and Farr RS: In
vitro acetylation of plasma proteins, enzymes and DNA by aspirin.
Nature. 219:68–69. 1968. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marchler-Bauer A, Derbyshire MK, Gonzales
NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI,
et al: CDD: NCBI's conserved domain database. Nucleic Acids Res.
43(Database Issue): D222–D226. 2015. View Article : Google Scholar
|
|
25
|
Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen
LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY, et al: Regulation of
G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and
cell survival during oxidative stress. EMBO J. 33:1304–1320.
2014.PubMed/NCBI
|
|
26
|
Choudhary C, Kumar C, Gnad F, Nielsen ML,
Rehman M, Walther TC, Olsen JV and Mann M: Lysine acetylation
targets protein complexes and co-regulates major cellular
functions. Science. 325:834–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Au SW, Gover S, Lam VM and Adams MJ: Human
glucose-6-phosphate dehydrogenase: The crystal structure reveals a
structural NADP (+) molecule and provides insights into enzyme
deficiency. Structure. 8:293–303. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jeffery J, Hobbs L and Jöernvall H:
Glucose 6-phosphate dehydrogenase from Saccharomyces cerevisiae:
Characterization of a reactive lysine residue labeled with
acetylsalicylic acid. Biochemistry. 24:666–671. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsouko E, Khan AS, White MA, Han JJ, Shi
Y, Merchant FA, Sharpe MA, Xin L and Frigo DE: Regulation of the
pentose phosphate pathway by an androgen receptor-mTOR-mediated
mechanism and its role in prostate cancer cell growth. Oncogenesis.
3:e1032014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang J, Yuan W and Chen Z, Wu S, Chen J,
Ge J, Hou F and Chen Z: Overexpression of G6PD is associated with
poor clinical outcome in gastric cancer. Tumour Biol. 33:95–101.
2012. View Article : Google Scholar
|
|
31
|
Du W, Jiang P, Mancuso A, Stonestrom A,
Brewer MD, Minn AJ, Mak TW, Wu M and Yang X: TAp73 enhances the
pentose phosphate pathway and supports cell proliferation. Nat Cell
Biol. 15:991–1000. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baba M, Yamamoto R, Iishi H, Tatsuta M and
Wada A: Role of glucose-6-phosphate dehydrogenase on enhanced
proliferation of pre-neoplastic and neoplastic cells in rat liver
induced by N-nitrosomorpholine. Int J Cancer. 43:892–895. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kuo W, Lin J and Tang TK: Human
glucose-6-phosphate dehydrogenase (G6PD) gene transforms NIH 3T3
cells and induces tumors in nude mice. Int J Cancer. 85:857–864.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kuo WY and Tang TK: Effects of G6PD
overexpression in NIH3T3 cells treated with tert-butyl
hydroperoxide or paraquat. Free Radic Biol Med. 24:1130–1138. 1998.
View Article : Google Scholar : PubMed/NCBI
|