Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an
inherited disorder with high penetrance, which approaches 100% with
increasing age (1). The disease
occurs with a prevalence of 2–3 per 100,000 in the population
(2). It is predominantly
characterized by tumors of the parathyroid glands,
gastroenteropancreatic tumors, pituitary adenomas, adrenal
adenomas, and neuroendocrine tumors of the thymus, lungs or
stomach, as well as non-endocrine lesions (2). The expression in terms of tumor
localization, age of onset and clinical aggressiveness, may vary
even between affected members of the same family. The clinical
manifestations of MEN1 are associated with the products of
secretion of the tumors rather than the primary sites or
metastases, and often appear at a young age (3). Management of MEN1 is based on
treatment or prevention of manifestations (4).
The syndrome is caused by inactivating mutations in
the tumor suppressor gene MEN1, coding for the 615-amino
acid protein menin (5).
MEN1 syndrome is inherited in an autosomal dominant
manner, which means that a single inherited mutation in the MEN1
gene predisposes to somatic loss of heterozygosity (LOH) during a
patient's lifetime. However, only once LOH has occurred does the
disease begin to develop. LOH predominantly occurs in the region at
which a mutation was inherited by the patient. This alteration may
occur in different tissues; however, a person bearing a single
mutation in MEN1 is certain to develop the disease. The
location, as well as the order and age of MEN1 manifestations are
unpredictable (3). The majority of
MEN1 mutations that have been found in affected families
result in truncated forms of menin. However, no genotype-phenotype
correlations have been proven (2).
In the present study, a kindred with a previously
unreported in-frame deletion in the MEN1 gene, with an
inheritance that is unexpected for Mendelian diseases was
described.
Materials and methods
Subjects and case history
A large Polish kindred was identified, in which 3
generations had MEN1 (Fig. 1). All
features presented within the case history occurred prior to the
commencement of the study, and all patients were enrolled during
the treatment stage. The index patient (II-3) was enrolled into the
study aged 50 with suspected MEN1. The patient underwent
parathyroidectomy due to primary hyperthyroidism at the age of 20.
Somatostatine-receptor scintigraphy showed pathological foci of
tracer uptake in the right mesogastrium projecting at the small
intestine loop, in the pancreatic tail and in both adrenal glands.
An abdominal magnetic resonance imaging scan confirmed the
pancreatic tail tumor with a size of 20×15×11 mm; a similar lesion
sized 16×13 mm was found in the topography of the inferior duodenal
flexure. These results were classified as typical for
neuroendocrine tumors. Ultrasound-guided fine-needle biopsy of the
pancreatic tumor showed well-differentiated neuroendocrine neoplasm
cells (NEN G2; Ki-67-3%). The patient did not consent to the
proposed neuroendocrine pancreatic tumor surgery (distal
pancreatectomy). The clinical course of the disease was stable with
unchanging tumor size and low chromogranin A levels until December
2014 when biochemical progression was observed (CgA-180 nml/l). The
patient did not turn up for further examination.
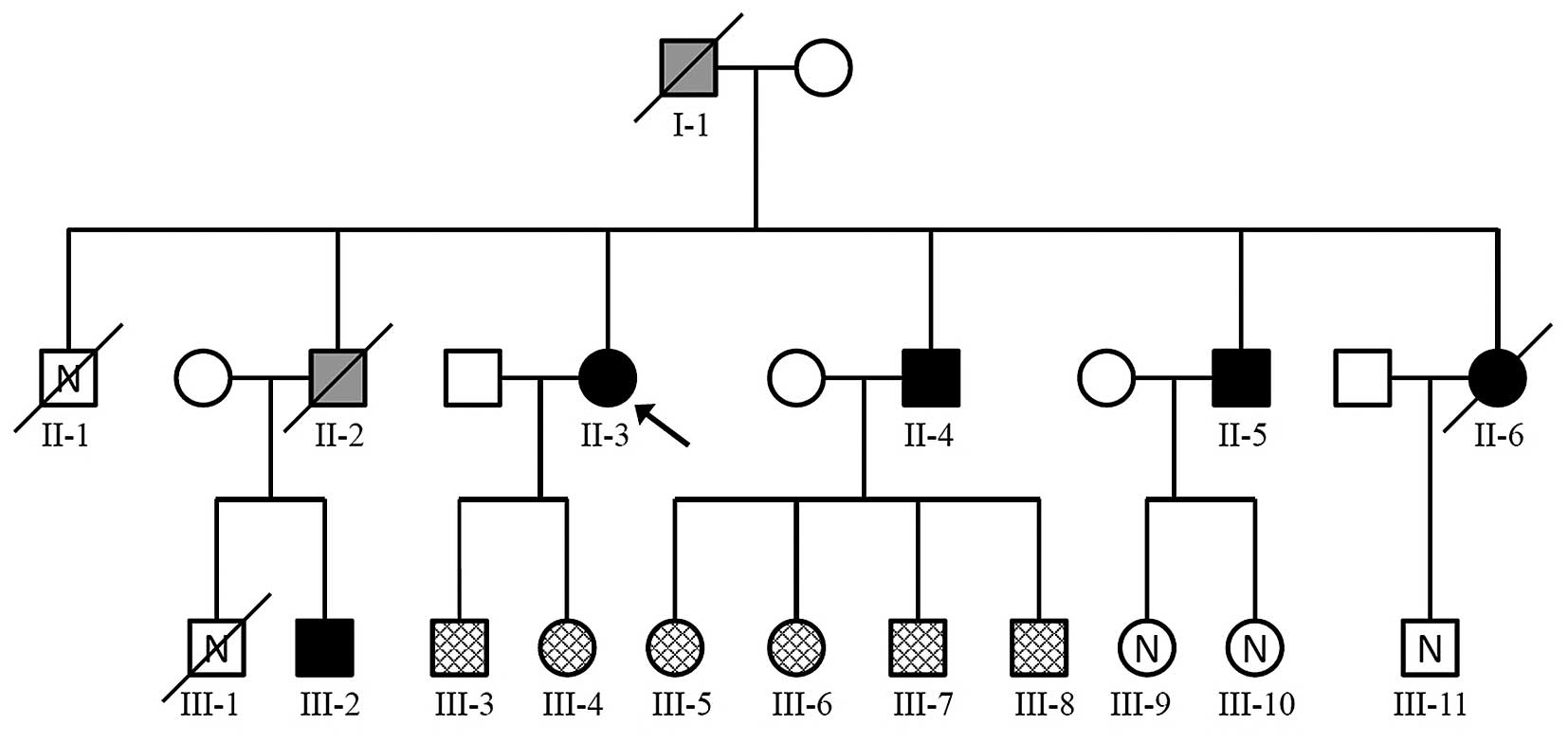 | Figure 1Pedigree showing MEN1 in a family. The
arrow indicates the index patient. Square, male; circle, female;
white, healthy individual, mutation status not tested; checked
pattern, healthy individual, Ala416del germline mutation absent;
Black, MEN1-affected individual with Ala416del germline mutation
detected. Grey, MEN1-affected individual, mutation status not
tested; /, dead; N, no data on health status. MEN1, multiple
endocrine neoplasia type 1. |
For the two children of the index patient-III-3 and
III-4 (enrolled for observation at age 24 and 22, respectively),
clinical observation, as well as diagnostic tests were
MEN1-negative.
Four of the index patient's siblings (II-2, II-4,
II-5 and II-6) also had symptoms of MEN1. The fifth sibling (II-1)
unexpectedly succumbed to mortality aged 24.
Their father (I-1) reportedly died at the age of 68
as a result of pancreatic head cancer. He also presented with
gastric ulcers, and underwent gastric resection ten years prior to
his death.
Case II-2 succumbed to hepatic encephalopathy at the
age of 38. In the past, he had presented with calcium-phosphate
disorder, potassium leakage and hepatitis type C. According to
this, the patient probably suffered from Cushing's syndrome. Of his
two sons, III-1 died a tragic death aged 15. The other (III-2) was
enrolled in our clinic aged 26 after parathyroidectomy due to
primary hyperthyroidism.
One of the surviving brothers (II-4) of the index
patient (aged 46) was diagnosed with primary hyperparathyroidism,
nephrolithiasis, tumors in both adrenal glands, and pancreatic
cancer. Computed tomography (CT) revealed mild hyperplasia of
adrenal glands. No typical changes for NET were observed in CT. He
was qualified for parathyroidectomy, but did not appear at set
appointments to continue therapy. The patient had four children:
III-5 aged 22, III-6 aged 21, III-7 aged 19, and III-8 aged 8, none
of the children presented with a clinical manifestation that
indicated MEN1.
The other brother (II-5) of the index patient was
enrolled in the study aged 34 with parathyroid adenoma and
hyperparathyroidism, after acute pancreatitis and after 3
extracorporeal shock wave lithotripsy surgeries. During early
puberty, the resection of a lipoma from the middle upper abdomen
was performed. The patient did not consent to surgery of the
parathyroid gland. There are no data regarding the health status of
the patient's two children (III-9 and III-10), aged 15 and 4.
The sister (II-6) of the index patient was enrolled
aged 37 with diffuse cancer due to a neuroendocrine tumor, most
probably from the pancreas, and also with recurrence of primary
hyperparathyroidism. She also suffered from nephrolithiasis,
euthyroid multinodular goiter and secondary diabetes. She succumbed
to hepatic encephalopathy aged 37. Her only son (aged 11) was
unavailable for enrollment in the present study.
All tested family members gave their written
informed consent for genetic testing. In the case of juvenile
members, additional consent was obtained from their legal
caretakers. The research has been approved by the local Ethics
Committee, (approval no. KBET/70/B/2013).
DNA isolation
Whole peripheral blood samples (2.6 ml) from each
patient were collected into EDTA-coated tubes (Sarstedt, Nümbrecht,
Germany). DNA was isolated with the QIAamp DNA Mini kit (Qiagen,
Hilden, Germany), according to the manufacturer's protocol.
A formalin-fixed paraffin-embedded (FFPE)
post-operative parathyroid gland was obtained from the index
patient. Seven sections, with a thickness of 10 μm each,
were cut from a region that contained ~70% cancerous tissue, as
assessed by light microscopy (Olympus BX51 with 40x UPlanFLN
eyepiece; Olympus Corporation, Tokyo, Japan), which involved the
fixation of the material in formalin, which was then processed by
the routine method and embedded in paraffin. Sections (4 μm)
were cut from paraffin blocks and stained with standard hematoxylin
and eosin (H&E; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) for histological examination. Corresponding paraffin cube
containing tumor tissue was selected on the basis of a comparison
with H&E slides. DNA from these sections was isolated using the
NucleoSpin FFPE DNA kit (Machery-Nagel, Dueren Germany). As a
negative somatic control, a mixture of two randomly selected
healthy post-operative FFPE parathyroid glands (which were removed
together with the thyroid during surgery for non-parathyroid
associated reasons from patients unrelated to the tested family and
negative for MEN1 syndrome) were used.
Sequencing
Amplification of products for
sequencing
The 9 coding exons of MEN1 (according to
transcript variant 1, RefSeq NM_000244.3; (ncbi.nlm.nih.gov/nuccore) were sequenced for the index
patient, her siblings and children, and only the one exon in which
the mutation was found, for the remaining participants. For PCR, 25
μl reaction mixtures with HotStarTaq polymerase (Qiagen)
were set up for each exon according to the standard recommendations
of the manufacturer. The mixtures contained 0.2 μM each of
the appropriate forward and reverse primer (Institute of
Biochemistry and Biophysics Polish Academy of Sciences, Warsaw,
Poland), and 100 ng DNA. Primers are listed in Table I. Reaction conditions: Initial
denaturation at 95°C for 15 min; 35 cycles including denaturation
at 95°C for 30 sec, annealing at 59°C for 30 sec, and elongation at
72°C for 30 sec; final elongation at 72°C for 10 min. Samples were
amplified in a Mastercycler realplex2 (Eppendorf, Hamburg,
Germany).
 | Table IPrimers used in polymerase chain
reaction. |
Table I
Primers used in polymerase chain
reaction.
| Amplified region | Primer
designation | Primer sequence | Product length
(bp) |
|---|
| Exon 2 | 2_F |
5′-AACCTTAGCGGACCCTGG-3′ | 654 |
| 2_R |
5′-ATAACACCTGCCGAACCTCA-3′ |
| Exon 3 | 3_F |
5′-CCCTTTCCCCATGTTAAAGC-3′ | 322 |
| 3_R |
5′-GGTGGCTTGGGCTACTACAG-3′ |
| Exon 4 | 4_F |
5′-CCTTTTCCTGGCTGTCATTC-3′ | 264 |
| 4_R |
5′-CCCACAGCAAGTCAAGTCTG-3′ |
| Exons 5–6 | 5–6_F |
5′-CTAAGGACCCGTTCTCCTCC-3′ | 322 |
| 5–6_R |
5′-CCTGCCTCAGCCACTGTTAG-3′ |
| Exon 7 | 7_F |
5′-GGCATTTGTGCCAGCAG-3′ | 261 |
| 7_R |
5′-GGAAACTGATGGAGGGGAAG-3′ |
| Exon 8 | 8_F |
5′-AGGTCCCTGGGGCTACC-3′ | 271 |
| 8_R |
5′-ATGGCCTGTGGAAGGGAG-3′ |
| Exon 9 | 9_F |
5′-CCCTCTGCTAAGGGGTGAG-3′ | 293 |
| 9_R |
5′-AAAAGTCTGACAAGCCCGTG-3′ |
| Exon 10 | 10_F |
5′-TCCTGGAGTTCCAGCCAC-3′ | 618 |
| 10_R |
5′-GAACATGGGCTCAGAGTTGG-3′ |
| External region
('ext') | 1f |
5′-ACCCAGAGCCAAGGTTCC-3′ | 79 |
| 2r |
5′-ATTTGCAGATGCCGTCGTAG-3′ |
| Inner region
wild-type allele ('wt') | Ww1 |
5′-AGGACCCTGAGTGCTTCGC-3′ | 54 |
| 2r |
5′-ATTTGCAGATGCCGTCGTAG-3′ |
| Inner region mutant
allele ('mut') | 1f |
5′-ACCCAGAGCCAAGGTTCC-3′ | 60 |
| Wm2 |
5′-GTAGAATCGCAGCAGGTĊGA-3′ |
Product purification and
visualization
The quality of the products was assessed by 2%
agarose electrophoresis in TAE buffer (Tris base, Thermo Fisher
Scientific, Inc.; acetic acid, Chempur, Piekary Slaskie, Poland;
EDTA, Avantor Performance Materials Poland S.A., Gliwice, Poland)
and visualized with ethidium bromide (Sigma-Aldrich, St. Louis, MO,
USA). The remaining PCR products were purified with the QIAquick
PCR Purification kit (Qiagen), according to the manufacturer's
protocol.
Sequencing PCR
The sequencing PCR reaction mixture included 1.25
μl BigDye Terminator v3.1 (Thermo Fisher Scientific, Inc.),
0.16 μM of the appropriate forward or reverse primer, and 20
ng of the appropriate purified product. PCR was conducted under
conditions recommended by the manufacturer. Specifically, PCR was
conducted in Mastercycler RealPlex2 (Eppendorf, Hamburg, Germany)
under the following conditions: Initial denaturation at 96°C for 1
min; 25 cycles including denaturation at 96°C for 10 sec, annealing
at 55°C for 5 sec and elongation at 60°C for 4 min.
Ethanol precipitation
To purify products after the sequencing PCR, 2
μl of 1.5 M sodium acetate/250 mM EDTA buffer, pH >8.0
were added to 10 μl of the reaction mixture. After
pipetting, 80 μl of 95% ethanol were added, the samples were
centrifuged for 15 min at 10,000 × g, and the supernatant
discarded. The pellets were washed with 75% ethanol and centrifuged
for 2 min in 10,000 × g. After dissolving the supernatants, DNA
pellets were left to air-dry, and dissolved in 20 μl
nuclease-free water (Ambion; Thermo Fisher Scientific, Inc.). The
whole procedure was conducted at room temperature. The purified
products were separated on the ABI3500 sequencer (Thermo Fisher
Scientific, Inc.).
Sequence analysis
The obtained sequences were aligned to the reference
NC_000011.10 with SeqScape software (version 2.7; Thermo Fisher
Scientific, Inc.). After identification of an exon-shortening
event, the overlapping sequence resulting from the heterozygous
deletion was analyzed manually using FinchTV (version 4.0;
Geospiza, Inc, Seattle, WA, USA).
Multiplex ligation-dependent probe
amplification (MLPA)
MLPA was performed with use of SALSA MLPA probemix
P017-C1, lot C1-0711 (MRC-Holland, Amsterdam, the Netherlands). The
reaction was performed according to the manufacturer's protocol,
using 100 ng DNA. Results were analyzed with Coffalyser.net (version 131123; MRC-Holland;
Amsterdam, Netherlands).
Testing for LOH
After an initial PCR with external primers, three
quantitative PCRs per sample were performed-with external primers
('ext'), and with internal primers specific for the wild-type
allele ('wt') or for the mutant allele ('mut'). Each sample was run
in triplicate. Primer sequences are presented in Table I.
At the time of primer design it was assured that the
tested patients and controls did not bear the rs2071313
polymorphism in germline material, in order to avoid lack of primer
binding due to this change.
The 10 μl real-time PCR mix contained 5
μl RT 2X PCR Master mix SYBR-C (A&A Biotechnology,
Gdynia, Poland), one of 4 successive ten-fold dilutions of the
external-PCR product, and standardized amounts of primers: 6
μM each for the 'wt' and the 'mut' reactions, and 3
μM forward and 6 μM reverse primer for 'ext'. The
reaction was set on ice; the prepared samples were put into the
heated thermal cycler. Reaction conditions were as follows: 40
cycles consisting of 95°C for 5 sec, 59°C for 10 sec, 72°C for 8
sec and 80°C for 15 sec; followed by a single step of 95°C for 15
sec. Fluorescence was measured after each 80°C step. A melting
curve analysis step was added after the reaction. Each sample was
run in triplicate. All reactions were run in the Mastercycler
realplex2 (Eppendorf, Hamburg, Germany).
The comparative Cq method was used to
evaluate the copy number in tested samples, with DNA from the index
patient as a reference ('IP blood'), which contains 1 wt and 1 mut
copy. Amplification efficiencies (E) were included into the
normalized copy number ratio equation E−ΔΔCQ (6). Average values and standard errors
were calculated from three independent experiments.
Results
DNA analysis
A heterozygous in-frame deletion, c.1246_1248delGCC,
was identified by sequencing the MEN1 gene for the index
patient, II-3. At the protein level, this leads to the deletion of
alanine at position 416 (p.Ala416del). According to available
variation databases, accessed on August 24, 2015 through the
Genome Browser at UCSC Genome Bioinformatics (7) and The Universal Mutation Database
(8), the identified mutation has
not been reported previously.
The index patient's two adult children, III-3 and
III-4 (at that time aged 24 and 22), did not exhibit any clinical
manifestation to suggest MEN1, and the absence of any variant in
MEN1 was confirmed by sequencing of the whole coding
region.
Sequence analysis revealed the presence of the
c.1246_1248delGCC mutation in the other affected family members,
II-4, II-5, II-6 and III-2. This mutation was not found in any of
the four asymptomatic children of patient II-4 (III-5, III-6, III-7
and III-8). MLPA was performed for all affected family members,
revealing no copy number changes among any exon or exon part of the
MEN1 gene. (data not shown).
In order to confirm that the detected
c.1246_1248delGCC mutation was causative of the disease in this
family, the post-operative FFPE parathyroid tissue from the index
patient (II-3) was analyzed for an additional, somatic MEN1
gene-function disrupting event (LOH), which typically occurs as a
large deletion in any region of the gene, but predominately on that
region of the wild-type allele in which the germline mutation
occurs on the other allele.
MLPA, although suggestive for a large deletion
encompassing the region with the mutation, gave ambiguous results,
most probably because of the poor quality of the DNA, reflected by
a poor Coffalyser Analysis Score for those samples.
The numbers of wild-type and mutated alleles were
determined by relative quantification. Average values of 3
independent experiments are shown in Fig. 2. As expected, in the blood sample
obtained from a healthy family member (the index patient's
daughter, 'CTRL blood') and in healthy parathyroid tissue ('CTRL
tissue') the number of wild-type alleles was twice that of the
index patient, and the mutated allele was absent. Results from the
transformed parathyroid tissue obtained from the index patient ('IP
tissue') revealed that the relative quantities of the wild-type and
the mutated allele were 0.33 and 1.81, respectively. If the LOH is
due to a copy loss without mutant allele duplication, these values
represent ~double of the factual amounts as, in fact, there is only
one remaining copy (the mutated) left in the sample. The finding of
0.33 wt copy numbers in the sample (where 0 wt copy numbers would
typically be expected) may arise from the surrounding tissue, which
may have a different number of wt copies, as FFPE slices containing
~70% tumor tissue were used for analysis.
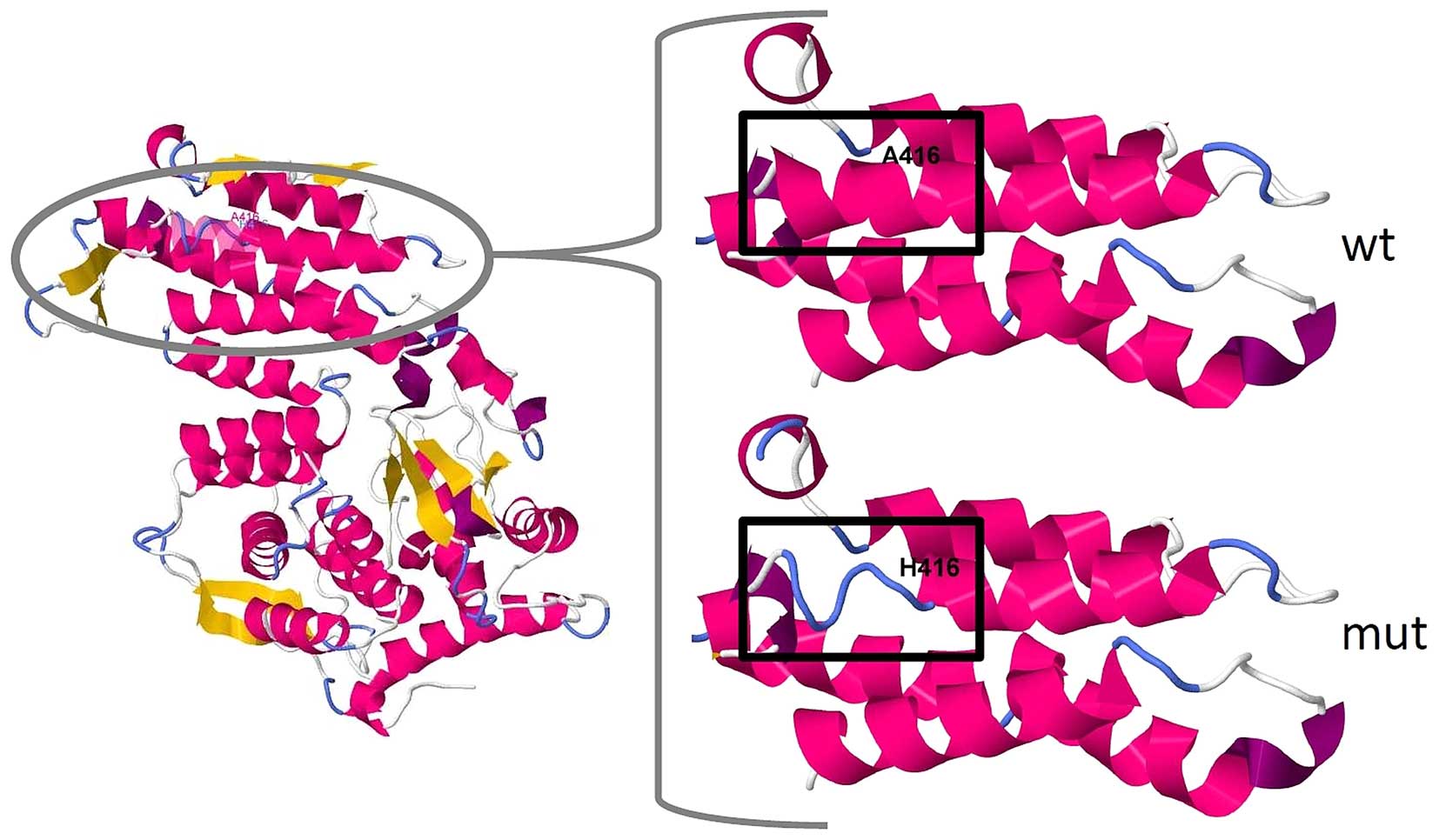
In silico analysis of the mutation
In the wild-type protein, alanine-416 is located in
an α-helix near the disordered structure of the menin protein.
According to SWISS-MODEL prediction (9), the deletion causes a disruption of
the N′-terminal end of this helix (Fig. 3). The tool PROVEAN marked
this mutation as 'deleterious' with a score of −10.97 (10). According to SIFT Indel, the
mutation is 'damaging' to the protein, with a confidence score of
0.894, which indicates that it affects a Pfam domain and that the
deletion is not located in a disordered region (11). Indeed, MEN1 is conserved in
bilateria (pfam05053), and Ala416 is located in a highly conserved
region of the protein (12).
Discussion
It has previously been demonstrated that genetically
diagnosed patients with MEN1 present with biochemical changes 10
years prior to the signs and symptoms of the disease (13), and an earlier diagnosis would allow
for more effective management of the disease. In addition, patients
who do not harbor a MEN1 mutation may be prevented from
undergoing unnecessary examination and lifelong surveillance
(14). However, predictive testing
can be offered to family members only after the disease-causing
nature of a variant has been unequivocally established (4). It is therefore of importance for the
patients and their family to confirm the pathogenic character of
their mutation in MEN1.
Large deletions and mutations at conserved donor and
acceptor splice sites, or mutations which introduce a premature
stop codon in the protein-coding region of MEN1 (nonsense
mutations and frameshift insertions or deletions) are explicitly
predicted to be disease causing (4). In the case of any other variant, it
has yet to be elucidated whether this is a pathogenic or a neutral
change. The present study provides evidence that the in frame
deletion c.1246_1248delGCC in the MEN1 gene, which, at the protein
level, leads to the deletion of alanine at position 416 in menin,
is a disease causing mutation. In order to confirm this finding,
the post-operative FFPE parathyroid tissue from the index patient
(II-3) was analysed for LOH in the region of the MEN1 gene. The
analysis of this large kindred resulted in a further notable
observation. In this family, either all or none of the siblings
inherited the disease. Statistically, this is not impossible, but
taking into account Mendelian inheritance patterns, this
observation may be noteworthy.
The present study is of importance as it
characterizes a newly discovered pathogenic mutation which may be
useful for any researcher or physician that encounters the same
mutation in another family or patient, but is unable to assess its
pathogenic status. In order to obtain more information about the
inheritance of this mutation, further investigations involving the
partners of the affected patients are required. The assessment of
environmental factor influence on the development of the disorder
would also be beneficial, however, for a small group of patients,
this kind of investigation may be difficult and have limited
statistical power.
Acknowledgments
This study was supported by the Polish Ministry of
Science and Higher Education (grant no. K/ZDS/003796). The authors
would like to thank Dr Łukasz Skalniak for his assistance in
experimental design. The authors would also like to thank the
described family for their collaboration.
References
|
1
|
Falchetti A: Genetic screening for
multiple endocrine neoplasia syndrome type 1 (MEN-1): When and how.
F1000 Med Rep. 2:142010.PubMed/NCBI
|
|
2
|
Lips CJM, Dreijerink KMA, van der Luijt
RB, van Nesselrooij BPM and Höppener JWM: MEN1.pdf. Genetic
Diagnosis of Endocrine Disorders. 1st edition. Weiss RE and
Refetoff S: Academic Press; pp. 261–270. 2010, View Article : Google Scholar
|
|
3
|
Lemos MC and Thakker RV: Multiple
endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations
reported in the first decade following identification of the gene.
Hum Mutat. 29:22–32. 2008. View Article : Google Scholar
|
|
4
|
Giusti F, Marini F and Brandi ML: Multiple
Endocrine Neoplasia Type 1. GeneReviews™. Pagon R: NIH; 2012
|
|
5
|
UniProt Consortium: Activities at the
universal protein resource (UniProt). Nucleic Acids Res.
42(Database Issue): D191–D198. 2014. View Article : Google Scholar :
|
|
6
|
Green MR and Sambrook J: Chapter 9.
Molecular Cloning: A Laboratory Manual. 2. 4th edition. CSH Press;
pp. 631–654. 2012
|
|
7
|
Kentw WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002.doi: 10.1101/gr.229102. Article
published online before print in May 2002.
|
|
8
|
Béroud C, Collod-Béroud G, Boileau C,
Soussi T and Junien C: UMD (Universal mutation database): A generic
software to build and analyze locus-specific databases. Hum Mutat.
15:86–94. 2000. View Article : Google Scholar
|
|
9
|
Arnold K, Bordoli L, Knopp J and Schwede
T: The SWISS-MODEL workspace: A web-based environment for protein
structure homology modelling. Bioinformatics. 22:195–201. 2006.
View Article : Google Scholar
|
|
10
|
Choi Y, Sims GE, Murphy S, Miller JR and
Chan AP: Predicting the functional effect of amino acid
substitutions and indels. PLoS One. 7:e466882012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu J and Ng PC: SIFT Indel: Predictions
for the functional effects of amino acid insertions/deletions in
proteins. PLoS One. 8:e779402013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marchler-Bauer A, Zheng C, Chitsaz F,
Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI,
Lanczycki CJ, et al: CDD: Conserved domains and protein
three-dimensional structure. Nucleic Acids Res. 41(Database Issue):
D348–D352. 2013. View Article : Google Scholar :
|
|
13
|
Lairmore TC, Piersall LD, DeBenedetti MK,
Dilley WG, Mutch MG, Whelan AJ and Zehnbauer B: Clinical genetic
testing and early surgical intervention in patients with multiple
endocrine neoplasia type 1 (MEN 1). Ann Surg. 239:637–645;
discussion 645–647. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lourenço DM Jr, Toledo RA, Coutinho FL,
Margarido LC, Siqueira SA, dos Santos MA, Montenegro FL, Machado MC
and Toledo SP: The impact of clinical and genetic screenings on the
management of the multiple endocrine neoplasia type 1. Clinics (Sao
Paulo). 62:465–476. 2007. View Article : Google Scholar
|