Introduction
The FLG gene, which encodes filament
aggregating protein (filaggrin), is located on human chromosome
1q21 (1). Filaggrin is a
filament-associated protein that binds to keratin fibers in
epithelial cells. Filaggrin monomers cluster into profilaggrin,
which is processed into filaggrin monomers by proteolysis.
Filaggrin is crucial for epidermal homeostasis and contributes to
the construction of the lipid envelope, which is critical for skin
barrier function (2). It is a
critical component of the stratum corneum, which provides primary
protection in humans due to its physical strength, hydration
status, skin pH and buffering capacity (3).
The importance of filaggrin in the frontline skin
barrier (4) is demonstrated by the
predisposition of individuals with filaggrin mutations to
various conditions, including dry skin, ichthyosis and atopic
dermatitis (5–7). Thus, it is necessary to fully
understand the functions of filaggrin to facilitate the treatment
of these diseases. It has been demonstrated that filaggrin
expression in keratinocytes results in decreased proliferation,
post-G1 phase arrest and loss of cell-cell adhesion
(8). In addition, filaggrin
increases the susceptibility of keratinocytes to apoptosis in
response to apoptosis-inducing stimuli (9). Furthermore, there is evidence to
suggest that filaggrin contributes to nuclear events associated
with apoptosis of epidermal keratinocytes (10). However, the effect of
filaggrin knockdown on the functions of normal human
epidermal keratinocytes (NHEKs) remains to be fully elucidated.
In the present study, the effect of filaggrin
absence on migration, invasion, adhesion, proliferation, apoptosis
and cell cycle progression in NHEKs was investigated. The results
of the present study may facilitate the determination of the
pathogenesis of filaggrin mutation-associated disorders.
Materials and methods
Cell culture
NHEKs were purchased from Invitrogen; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA), and cultured in
EpiLife® medium supplemented with growth factors
(Invitrogen; Thermo Fisher Scientific, Inc.). Cells were cultured
in 10 cm dishes in a 5% CO2 incubator at 37°C. The
medium was replaced every second day and the cells were split 1:2
every 3 days. For experiments other than cell proliferation and
adhesion, cells were cultured with 1.5 mM calcium for 24 h to
induce differentiation.
Filaggrin silencing by LV infection
The present study used the following LV-encoding
shRNA infection to knockdown filaggrin: GTTGGCTCAAGCA
TATTATTT (position: nt-274). The negative control (NC) shRNA
sequence was CAACAAGATGAAGAGCACC. The complementary DNA of the
shRNA was inserted into the LV gene transfer vector and the double
stranded shRNA oligo was cloned into pGLV-H1-GFP (Shanghai
GenePharma Co., Ltd., Shanghai, China) with BamHI and
EcoRI (Thermo Fisher Scientific, Inc.). The construct was
validated by western blotting. The shRNA-infected cells were
referred to as the LV group, cells infected with control
non-filaggrin shRNA as the NC group, and cells without
infection as the blank group.
The constructs were diluted 1:4 with
EpiLife® medium containing 10% fetal calf serum (FCS;
Invitrogen; Thermo Fisher Scientific, Inc.) and 10 mg/ml
polybrene® (Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), to a final concentration of 5 µg/ml; this was the LV
working solution. When the NHEKs reached 90% confluence, the cells
were digested using 1 ml 0.25% trypsin-ethylenediaminetetraacetic
acid (EDTA) solution, and single cell suspensions were prepared.
Cells were seeded into 6-well plates (1×106 cells/well)
and incubated in EpiLife® medium containing 10% FCS at
37°C and 5% CO2 for 24 h. Following this, the
EpiLife® medium was removed and 1 ml LV working solution
was added to each experimental well and incubated for 24 h. Cells
were observed under a fluorescence microscope (Olympus America,
Inc., Melville, NY, USA). The results of the preliminary
experiments revealed that LV was stably expressed for four
days.
Migration assays
Cell migration was analyzed using Transwell inserts
with an 8-µm pore membrane (BD Biosciences, San Jose, CA,
USA) as described previously (11,12).
The LV-infected cells were grown to sub-confluence (75–80%) and
then serum-starved for 24 h. Following detachment with trypsin, the
cells were washed with phosphate-buffered saline (PBS) and
resuspended in serum-free medium. Subsequently, 100 µl cell
suspension (3×105 cells/ml) was added to the upper
chamber. The membranes were coated with 0.01% collagen type I in
0.01 N HCl (Sigma-Aldrich, St. Louis, MO, USA). The lower chamber
was filled with 700 µl RPMI-1640 medium (Invitrogen; Thermo
Fisher Scientific, Inc.) with 15% FCS. Following a 48-h incubation
in a 5% CO2 incubator at 37°C, membranes were removed.
Cells remaining on the upper side of the membranes were wiped off
using cotton swabs, while cells that had migrated to the lower
chamber were fixed with 500 µl methanol for 10 min at −20°C
and stained with 200 µl 0.1% crystal violet for 30 min at
37°C. Images of five separate fields selected at random
(magnification, ×100) were captured from each well and the number
of migrated cells was counted. The mean number of migrated cells
per field was calculated for each experimental condition.
Cell adhesion and proliferation
assay
Flat-bottom culture plates (96-well) were coated
with 60 µl of Matrigel diluted 1:5 in serum-free
EpiLife® medium, incubated in 5% CO2 at 37°C
for 4 h. NC, blank and LV-treated cells were harvested with 1 ml
trypsin-EDTA solution 48 h following LV infection, washed twice
with PBS and resuspended in EpiLife® medium. Cells were
added to the coated 96-well plates (5×104 cells/well) in
quintuplicate and incubated at 37°C for 3 h. Subsequently, the
96-well plates were washed twice with PBS to remove unbound cells
and 100 µl fresh medium was then added to each well. The
remaining adhesive cells in the plate were assessed using a Cell
Counting kit (CCK)-8 (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) according to the manufacturer's instructions.
Finally, 96-well plates were examined at 450 nm using a plate
reader 2.5 h later. The results were calculated with the following
formula: Adhesion rate=[mean optical density (OD) of treated
cells]/(mean OD of corresponding control) ×100.
In addition, the cell proliferation assay was
performed with the CCK-8 (13).
Cells were seeded in 96-well plates at ~5×103 cells/well
and cultured in EpiLife® medium at 37°C. At the
indicated time points (0, 24, 48, 72 and 96 h), 10 µl CCK-8
solution was added to each well and incubated for 2.5 h, followed
by examination at 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA.).
Cell apoptosis assays
A total of 72 h following LV infection, cells were
harvested with EDTA free-trypsin for apoptosis analysis or passaged
and cultured at a density of 5×105 cells/ml in a 6-well
plate for 48 h prior to apoptosis analysis. Analysis was performed
using an annexin V-phycoerythrin (PE)/7-aminoactinomycin D (7-AAD)
apoptosis detection kit (BD Biosciences) according to the
manufacturer's instructions (14).
Briefly, following washing twice with cold PBS and centrifugation
at 300 x g for 5 min at 4°C, the cells were resuspended in 50
µl binding buffer and 5 µl 7-AAD for 15 min at room
temperature in the dark. Cells were then incubated with 450
µl binding buffer and 1 µl annexin V-PE for 15 min in
the dark and detected immediately on a FACSCalibur flow cytometer
(BD Biosciences) using CellQuest software version 3.2 (BD
Biosciences).
Cell cycle analysis
Following LV infection, cells were incubated at a
density of 5×105 cells/ml in a 6-well plate for 48 h.
Cells (1–5×106 cells/ml) were then washed twice with
ice-cold PBS, resuspended in 500 µl PBS and fixed with 1.5
ml of precooled 100% ethanol overnight at 4°C. Following two PBS
washes, the fixed cells were centrifuged (300 x g, 5 min, 4°C) to
remove the ethanol. Cells were adjusted to 1–10×105
cells/ml and incubated with 150 µl RNaseA (250–500
µg/ml) for 30 min at 37°C, followed by the addition of 100
µl propidium iodide (Sigma-Aldrich) for 30 min at 4°C in the
dark. DNA content was analyzed using a FACSCalibur flow cytometer
at an excitation wavelength of 488 nm (15).
Western blotting
Cells were harvested 72 h following LV infection and
washed twice with ice-cold PBS. Briefly, the cells were lysed and
homogenized with radioimmunoprecipitation assay lysis buffer
(Thermo Fisher Scientific, Inc.) for 30 min on ice. Samples were
diluted with 2X SDS-PAGE loading buffer (1:1), followed by thermal
denaturation at 100°C for 5 min. Following cooling, the
supernatants were collected by centrifugation at 10,000 x g at 4°C
for 10 min. The protein concentration was quantified by a
bicinchoninic acid assay (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Protein samples (30
mg) were separated using a 10% SDS-PAGE gel (100 mV), and
transferred to nitrocellulose membranes. Following blocking with 5%
nonfat milk in Tris-buffered saline and Tween 20 (TBST) for 2 h,
the membranes were incubated overnight at 4°C with primary
antibodies: Rabbit anti-filaggrin monoclonal antibody (catalog no.
PRB-417P; 1:250) purchased from Covance, Inc. (Princeton, NJ, USA)
and rabbit anti-GAPDH antibody (catalog no. sc-25778; 1:3,000)
purchased from Santa Cruz Biotechnology, Inc. Membranes were washed
with TBST three times and incubated with a goat-rabbit IgG
secondary antibody conjugated to horseradish peroxidase (catalog
no. sc-2030; 1:1,000) purchased from Santa Cruz Biotechnology, Inc.
Reactive bands were detected by enhanced chemiluminescence reagent
(Pierce; Thermo Fisher Scientific, Inc.). Protein expression levels
were quantified using Gel-Pro Analyzer software version 3.1 (Media
Cybernetics, Inc., Rockville, MD, USA). GAPDH served as the
internal reference.
Statistical analysis
Data are presented as the mean ± standard deviation,
and all statistical analyses were conducted using SPSS version 13.0
(SPSS, Inc., Chicago, IL, USA). Differences between the
experimental groups were analyzed using Student's t-test, or
one-way analysis of variance followed by the least significant
difference post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
shRNA infection effectively knocks down
filaggrin expression levels
The effect of shRNA infection on the protein
expression levels of filaggrin was determined by western blotting.
As presented in Fig. 1, shRNA
infection resulted in a significant decrease in filaggrin protein
expression levels at 72 h (P=0.008 vs. NC group). The results of
the present study indicated that filaggrin was successfully
knocked down.
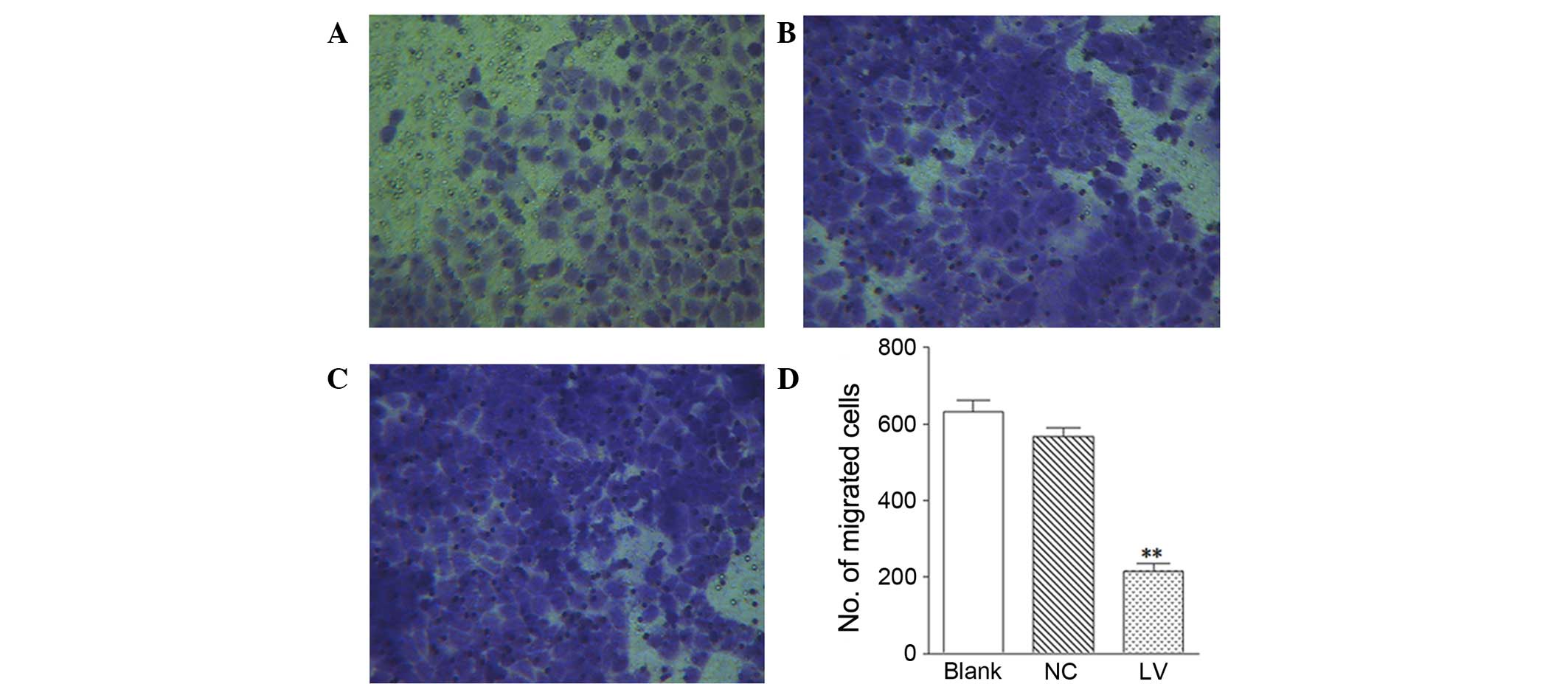
Filaggrin knockdown inhibits cell
migration
The impact of filaggrin knockdown on cell
migration was investigated using Transwell inserts. As presented in
(Fig. 2A–D), the LV group
(Fig. 2A) had significantly less
migrated cells than the NC (Fig.
2B; P=0.0059) and blank groups (Fig. 2C). This observation suggested that
a lack of filaggrin may markedly inhibit the migration of
NHEKs.
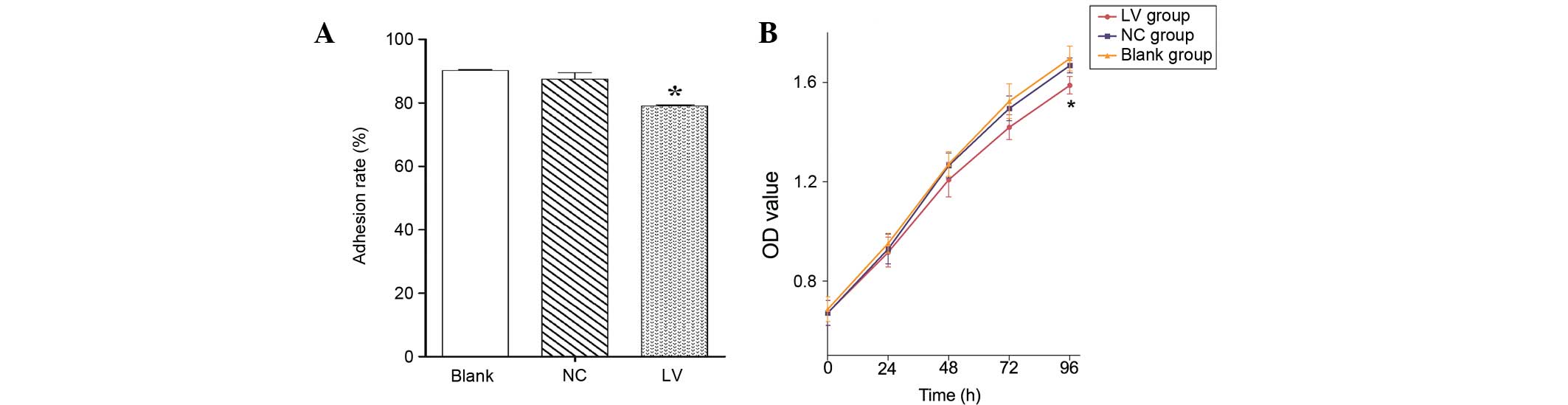
Filaggrin knockdown suppresses cell
adhesion and proliferation
In addition to cell migration and invasion, the role
of filaggrin in cell adhesion and proliferation was
analyzed. As presented in Fig. 3A,
adhesion of the LV group was significantly inhibited compared with
the NC (P=0.023) and blank groups. Furthermore, filaggrin
knockdown had no significant influence on cell proliferation at 72
h; however, a significant decrease was observed at 96 h in the LV
group compared with the NC (P=0.034) and blank groups (Fig. 3B).
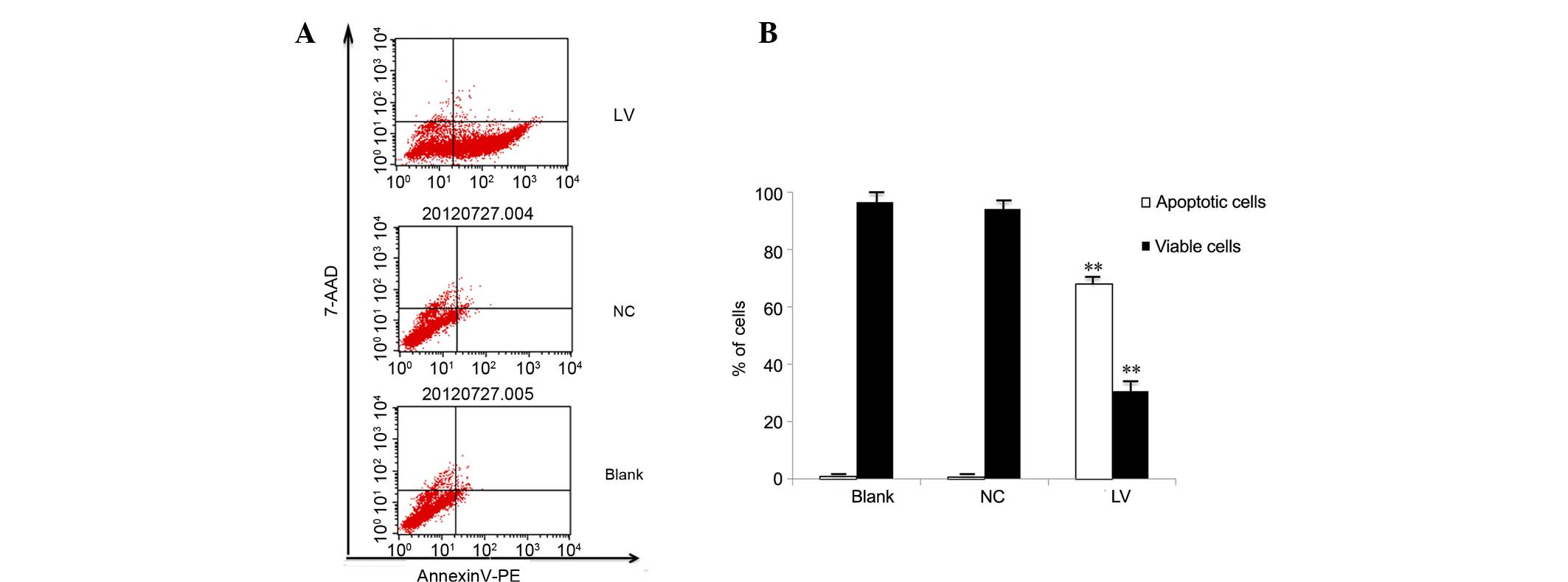
Filaggrin knockdown induces
apoptosis
An annexin V-PE/7-AAD apoptosis detection kit was
used to determine the effect of filaggrin knockdown on
apoptosis. As presented in Fig. 4,
the proportion of early apoptotic cells was significantly increased
in the LV group compared with the NC and blank groups (68.01 vs.
0.76 and 0.92%, respectively; P=0.0065 vs. NC group), while the
proportion of viable cells was significantly decreased (30.60 vs.
94.00 and 96.6%; P=0.0063 vs. NC group), at 72 h following
infection. Passaged cells (72 h following infection and 48 h
following passage), exhibited a similarly significant increase in
the percentage of early apoptotic cells (72.14 vs. 0.56 and 0.82%;
P=0.0054 vs. NC group) and a significant decrease in the proportion
of viable cells (25.88 vs. 94.22 and 95.23%; P=0.0076 vs. NC group;
Fig. 5). Therefore,
filaggrin knockdown appeared to induce apoptosis of
NHEKs.
Filaggrin knockdown alters cell cycle
progression
Flow cytometry was performed to evaluate the effect
of filaggrin knockdown on cell cycle progression in NHEKs.
As presented in Fig. 6A–C, the
cell cycle distribution pattern of the LV group was distinct to
that of the NC and blank groups. The proportion of cells in S phase
was significantly reduced (18.19 vs. 25.90 and 26.07%; P=0.034 vs.
NC group); however, the proportion of cells in G1 and
G2 phases was significantly increased in the LV group
compared with the NC and Blank groups (71.82 vs. 65.29 and 66.25%;
P=0.031 vs. NC group; and 3.47 vs. 7.92 and 6.89%; P=0.0064 vs. NC
groups). The increase in cells that had undergone filaggrin
knockdown in G1 and G2 stages suggested that
G1/S transition was inhibited and S/G2
transition accelerated as a result of filaggrin
knockdown.
Discussion
Filaggrin mutations may contribute to various
diseases, including dry skin, ichthyosis vulgaris, atopic eczema
and atopic dermatitis (16).
Filaggrin is crucial for epidermal homeostasis and differentiation,
and skin barrier function (17).
The present study aimed to investigate the effects of
filaggrin knockdown on various functions of NHEKs, including
cell migration, invasion, adhesion, proliferation, apoptosis and
cell cycle progression. Filaggrin was successfully knocked
down in NHEKs by infection with LV encoding shRNA of
filaggrin, resulting in inhibition of cell migration,
adhesion and proliferation, promotion of apoptosis, and disturbance
of cell cycle progression.
Cell migration is a highly integrated multistep
process, which contributes to tissue formation, regeneration and
remodeling, wound healing, and the immune response (18). Tissue repair, regeneration and
remodeling require active cell motility, which is also reliant on
cell adhesion (19). In addition,
cell adhesion is involved in the maintenance of multicellular
structures, signal transduction and cancer metastasis (20). Cell migration and adhesion are
closely associated with terminal differentiation and epidermal
homeostasis (21,22). Our previous study demonstrated that
filaggrin knockdown inhibits expression of epidermal
differentiation-associated proteins (23). Consistent with these findings, the
present study revealed suppression of NHEK migration and adhesion
as a result of filaggrin knockdown. Therefore, the absence
of filaggrin may prohibit epidermal differentiation by
suppressing the migration and adhesion of NHEKs.
Furthermore, Filaggrin knockdown inhibited
cell proliferation and promoted apoptosis. Cell proliferation and
apoptosis are involved in epidermal homeostasis and skin wound
repair (21,24). These observations indicated that
the absence of filaggrin may deregulate epidermal
homeostasis and delay wound healing by inhibiting NHEK migration,
adhesion and proliferation, and promoting NHEK apoptosis.
Cell cycle progression in the present study was also
altered by filaggrin knockdown, as indicated by the
increased proportions of cells in G1 and G2
phases, and the reduced proportions of cells in S phase. Therefore,
filaggrin absence slowed down G1/S transition and
accelerated S/G2 transition, which may provide an
explanation for the inhibition of proliferation by filaggrin
knockdown. These results suggest that filaggrin is involved
in the regulation of cell cycle progression; however, further
studies are required to validate this effect.
Evidence suggests that the MAPK and phosphoinositide
3-kinase/Akt signaling pathways are involved in regulating
keratinocyte differentiation, proliferation and apoptosis (25–28).
Downregulation of Akt has been reported to promote apoptosis, and
inhibit cell migration and proliferation (29). Activation of Akt reverses cell
cycle arrest in G1 and G2 phases in response
to DNA injury (30). Furthermore,
the effect of Akt on cellular survival and metabolism is mediated
by binding to downstream NF-κB (31). Filaggrin absence may inhibit
the differentiation of NHEKs by suppressing phosphorylation of P38,
ERK1/2, JNK, Akt and NF-κB. Therefore, filaggrin knockdown
may inhibit cell migration, adhesion and proliferation, promote
cell apoptosis and disturb cell cycle progression via suppression
of these signaling pathways. However, further experiments are
required to confirm this hypothesis.
In conclusion, the results of the present study
demonstrate that filaggrin knockdown inhibits NHEK
migration, adhesion and proliferation, promotes apoptosis and
disturbs cell cycle progression. These findings contribute to the
understanding of the role of filaggrin in epidermal
keratinocytes, and may facilitate the determination of the
pathogenesis of filaggrin mutation-associated disorders.
Acknowledgments
The present study was supported by the China
Postdoctoral Science Foundation (grant nos. 2014M550370 and
2015T80740).
References
|
1
|
McGrath JA and Uitto J: The filaggrin
story: Novel insights into skin-barrier function and disease.
Trends Mol Med. 14:20–27. 2008. View Article : Google Scholar
|
|
2
|
Ovaere P, Lippens S, Vandenabeele P and
Declercq W: The emerging roles of serine protease cascades in the
epidermis. Trends Biochem Sci. 34:453–463. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Levin J, Friedlander SF and Del Rosso JQ:
Atopic dermatitis and the stratum corneum: Part 1: The role of
filaggrin in the stratum corneum barrier and atopic skin. J Clini
Aesthet Dermatol. 6:16–22. 2013.
|
|
4
|
Sandilands A, Sutherland C, Irvine AD and
McLean WH: Filaggrin in the frontline: Role in skin barrier
function and disease. J Cell Sci. 122:1285–1294. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weidinger S, Illig T, Baurecht H, Irvine
AD, Rodriguez E, Diaz-Lacava A, Klopp N, Wagenpfeil S, Zhao Y, Liao
H, et al: Loss-of-function variations within the filaggrin gene
predispose for atopic dermatitis with allergic sensitizations. J
Allergy Clin Immunol. 118:214–219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O'Regan GM, Sandilands A, McLean WH and
Irvine AD: Filaggrin in atopic dermatitis. J Allergy Clin Immunol.
124(3 Suppl 2): R2–R6. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun LD, Xiao FL, Li Y, Zhou WM, Tang HY,
Tang XF, Zhang H, Schaarschmidt H, Zuo XB, Foelster-Holst R, et al:
Genome-wide association study identifies two new susceptibility
loci for atopic dermatitis in the Chinese Han population. Nat
Genet. 43:690–694. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Presland RB, Kuechle MK, Lewis SP,
Fleckman P and Dale BA: Regulated expression of human filaggrin in
keratinocytes results in cytoskeletal disruption, loss of cell-cell
adhesion, and cell cycle arrest. Exp Cell Res. 270:199–213. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kuechle MK, Presland RB, Lewis SP,
Fleckman P and Dale BA: Inducible expression of filaggrin increases
keratinocyte susceptibility to apoptotic cell death. Cell Death
Differ. 7:566–573. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ishida-Yamamoto A, Tanaka H, Nakane H,
Takahashi H, Hashimoto Y and Iizuka H: Programmed cell death in
normal epidermis and loricrin keratoderma. Multiple functions of
profilaggrin in keratinization. J Investig Dermatol Symp Proc.
4:145–149. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang J, Shan L, Koussih L, Redhu NS,
Halayko AJ, Chakir J and Gounni AS: Pentraxin 3 (PTX3) expression
in allergic asthmatic airways: Role in airway smooth muscle
migration and chemokine production. PLoS One. 7:e349652012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kramer N, Walzl A, Unger C, Rosner M,
Krupitza G, Hengstschläger M and Dolznig H: In vitro cell migration
and invasion assays. Mutat Res. 752:10–24. 2013. View Article : Google Scholar
|
|
13
|
Kong X, Chang X, Cheng H, Ma R, Ye X and
Cui H: Human epididymis protein 4 inhibits proliferation of human
ovarian cancer cells via the mitogen-activated protein kinase and
phosphoinositide 3-kinase/AKT pathways. Int J Gynecol Cancer.
24:427–436. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xu QF, Pan YW, Li LC, Zhou Z, Huang QL,
Pang JC, Zhu XP, Ren Y, Yang H, Ohgaki H and Lv SQ: MiR-22 is
frequently downregulated in medulloblastomas and inhibits cell
proliferation via the novel target PAPST1. Brain Pathol.
24:568–583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Choi YK, Seo HS, Choi HS, Choi HS, Kim SR,
Shin YC and Ko SG: Induction of Fas-mediated extrinsic apoptosis,
p21WAF1-related G2/M cell cycle arrest and ROS generation by
costunolide in estrogen receptor-negative breast cancer cells,
MDA-MB-231. Mol Cell Biochem. 363:119–128. 2012. View Article : Google Scholar
|
|
16
|
Thyssen JP, Ross-Hansen K, Johansen JD,
Zachariae C, Carlsen BC, Linneberg A, Bisgaard H, Carson CG,
Nielsen NH, Meldgaard M, et al: Filaggrin loss-of-function mutation
R501X and 2282del4 carrier status is associated with fissured skin
on the hands: Results from a cross-sectional population study. Br J
Dermatol. 166:46–53. 2012. View Article : Google Scholar
|
|
17
|
McAleer MA and Irvine AD: The
multifunctional role of filaggrin in allergic skin disease. J
Allergy Clin Immunol. 131:280–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ridley AJ, Schwartz MA, Burridge K, Firtel
RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR: Cell
migration: Integrating signals from front to back. Science.
302:1704–1709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Griffith LG and Naughton G: Tissue
engineering-current challenges and expanding opportunities.
Science. 295:1009–1014. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gumbiner BM: Cell adhesion: The molecular
basis of tissue architecture and morphogenesis. Cell. 84:345–357.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livshits G, Kobielak A and Fuchs E:
Governing epidermal homeostasis by coupling cell-cell adhesion to
integrin and growth factor signaling, proliferation, and apoptosis.
Proc Natl Acad Sci USA. 109:4886–4891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Amelio I, Lena AM, Viticchiè G,
Shalom-Feuerstein R, Terrinoni A, Dinsdale D, Russo G, Fortunato C,
Bonanno E, Spagnoli LG, et al: miR-24 triggers epidermal
differentiation by controlling actin adhesion and cell migration. J
Cell Biol. 199:347–363. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dang NN, Pang SG, Song HY, An LG and Ma
XL: Filaggrin silencing by shRNA directly impairs the skin barrier
function of normal human epidermal keratinocytes and then induces
an immune response. Braz J Med Biol Res. 48:39–45. 2015. View Article : Google Scholar :
|
|
24
|
Lewis CJ, Mardaryev AN, Poterlowicz K,
Sharova TY, Aziz A, Sharpe DT, Botchkareva NV and Sharov AA: Bone
morphogenetic protein signaling suppresses wound-induced skin
repair by inhibiting keratinocyte proliferation and migration. J
Invest Dermatol. 134:827–837. 2014. View Article : Google Scholar
|
|
25
|
Popp T, Egea V, Kehe K, Steinritz D,
Schmidt A, Jochum M and Ries C: Sulfur mustard induces
differentiation in human primary keratinocytes: Opposite roles of
p38 and ERK1/2 MAPK. Toxicol Lett. 204:43–51. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Efimova T, Broome AM and Eckert RL: A
regulatory role for p38 delta MAPK in keratinocyte differentiation.
Evidence for p38 delta-ERK1/2 complex formation. J Bioll Chem.
278:34277–34285. 2003. View Article : Google Scholar
|
|
27
|
Jonak C, Mildner M, Klosner G, Paulitschke
V, Kunstfeld R, Pehamberger H, Tschachler E and Trautinger F: The
hsp27kD heat shock protein and p38-MAPK signaling are required for
regular epidermal differentiation. J Dermatol Sci. 61:32–37. 2011.
View Article : Google Scholar
|
|
28
|
Calautti E, Li J, Saoncella S, Brissette
JL and Goetinck PF: Phosphoinositide 3-kinase signaling to Akt
promotes keratinocyte differentiation versus death. J Bioll Chem.
280:32856–32865. 2005. View Article : Google Scholar
|
|
29
|
Pap M and Cooper GM: Role of glycogen
synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell
survival pathway. J Bioll Chem. 273:19929–19932. 1998. View Article : Google Scholar
|
|
30
|
Kandel ES, Skeen J, Majewski N, Di
Cristofano A, Pandolfi PP, Feliciano CS, Gartel A and Hay N:
Activation of Akt/protein kinase B overcomes a G(2)/M cell cycle
checkpoint induced by DNA damage. Mol Cell Biol. 22:7831–7841.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Faissner A, Heck N, Dobbertin A and
Garwood J: DSD-1-Proteoglycan/Phosphacan and receptor protein
tyrosine phosphatase-beta isoforms during development and
regeneration of neural tissues. Adv Exp Med Biol. 557:25–53. 2006.
View Article : Google Scholar : PubMed/NCBI
|