Introduction
Non-esterified free fatty acids (FFAs) impose
lipotoxic effects on the kidney (1) by increasing production of reactive
oxygen species (ROS) (2), thus
leading to mitochondrial dysfunction (3). Although obesity is preventable, many
people use smoking as a means to lose weight (4,5).
Nicotine (NIC) is a major component of tobacco smoke (6) and E-cigarettes (7), and is responsible for the association
between smoking and kidney injury (8) through increases in oxidative stress
(9). Therefore, it is possible
that FFA- and NIC-associated oxidative stresses are superimposed,
resulting in enhanced oxidative stress in the kidneys of obese
smokers. A previous in vivo study revealed that NIC exposure
exacerbates high-fat diet (HFD)-associated FFA deposition and
oxidative stress in mouse kidneys (10).
It has previously been reported that NIC and oleic
acid (OA) increase the transcription of the pro-oxidant p66shc
gene, resulting in elevated mitochondrial ROS production and
consequent mitochondrial depolarization-dependent injury in
cultured renal proximal tubule cells (11,12).
Our recent study demonstrated that NIC exposure increased oxidative
stress with a concomitant increase in p66shc expression in the
kidneys of mice fed a HFD compared with those fed a normal diet
(10). Therefore, it is possible
that NIC exacerbates FFA-mediated oxidative stress, in part, due to
additive effects on p66shc. Furthermore, oxidative stress is not
solely the result of excessive production of ROS; it may also be
due to impaired activation of the antioxidant defense system
(13). Our previous study revealed
that while HFD increases expression of the antioxidant manganese
superoxide dismutase (MnSOD), its expression was reduced in the
kidneys of mice exposed to NIC (10). Notably, p66shc is important for the
direct production of mitochondrial ROS (11,12)
and the suppression of MnSOD in non-renal cells (14–16).
Therefore, the present study hypothesized that NIC
exposure would exacerbate renal oxidative stress and consequent
renal lipotoxicity via a p66shc-dependent increase in ROS
production and suppression of MnSOD expression in renal proximal
tubule cells.
Materials and methods
Cell line and treatment
The NRK52E renal proximal tubule cell line was
purchased from American Type Culture Collection (Manassas, VA, USA)
and maintained in Dulbecco's modified Eagle's medium supplemented
with 10% fetal bovine serum (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C in an atmosphere containing 5%
CO2. Cells were treated with 200 µM NIC
(Sigma-Aldrich, St. Louis, MO, USA), 100 µM OA
(Sigma-Aldrich), or with a combination of NIC and OA.
Modulation of p66shc and MnSOD
expression
NRK52E cells were transfected with a p66shc
expression plasmid to overexpress (OE) or a short hairpin (sh)
p66shc plasmid to knockdown (k.d.) p66shc expression (17,18)
using Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. These
plasmids were prepared in our laboratory. In order to suppress
MnSOD expression, NRK52E cells were transfected with a shMnSOD
plasmid (Addgene, Inc., Cambridge, MA, USA) using
Lipofectamine® 3000 transfection reagent, according to
the manufacturer's protocol. Cells were transfected with the
aforementioned plasmids prior to treatment.
Determination of intracellular ROS
production
NRK52E cells were cultured in T25 flasks and
transfected with the aforementioned plasmids. Following
trypsinization, cells were counted and loaded with the
oxidant-sensitive dye 2′,7′-dichlorofluorescein-diacetate (100
µM; Thermo Fisher Scientific, Inc.) as previously described
(17). The cells were incubated
for 30 mins at 37°C and the dye was washed away with fresh Hanks'
balanced salt solution (Sigma-Aldrich). The cells were then seeded
at a density of 0.2×106 cells/well and were treated with
200 µM NIC, 100 µM OA, or with a combination of NIC
and OA for 24 h. ROS production was determined by recording the
increase in fluorescence at 485 nmexc/530
nmem in 30-min-intervals for up to 120 min using a
microplate reader (FluoroCount, Packard; Molecular Devices, LLC,
Sunnyvale, CA, USA). ROS production was calculated as: Change in
fluorescence / 30 min / 0.2×106 cells, and was expressed
as a percentage of the corresponding untreated values.
Determination of cell injury
The extent of cell injury was determined using the
fluorescent CytoTox-ONE™ Homogenous Membrane Integrity assay kit
(Promega Corporation, Madison, WI, USA), according to the
manufacturer's protocol. Briefly, cells cultured in 96-well plates
were transfected/treated as required and lactate dehydrogenase
(LDH) content in the supernatant was compared with total cellular
LDH content. LDH release was calculated as percentage of total LDH
content (19). In some
experiments, cells were pretreated with 10 µM
N-acetylcysteine (NAC: Sigma-Aldrich) for 30 min prior to treatment
with NIC and OA.
Reporter luciferase assay
NRK52E cells were cultured in 24-well plates and
were transfected with the following reporter luciferase plasmids:
p66shc-promoter luciferase (20)
provided by Dr Irani (Cardiovascular Institute, University of
Pittsburgh Medical Center, Pittsburgh, PA, USA), MnSOD
promoter-reporter-luciferase (21)
or a luciferase plasmid that harbors 6 canonical forkhead box
(FOXO) binding sites (6xDBE) to determine FOXO-dependent
transcription (22) provided by Dr
Burgering (Department of Molecular Cancer Research, University
Medical Center Utrecht, Utrecht, Netherlands) together with
Renilla luciferase (Promega Corporation) using
Lipofectamine® 3000 reagent. Firefly and Renilla
luciferase activity levels were determined after 24 h using the
Dual Luciferase assay kit (Promega Corporation). Luciferase
activity levels were calculated as a ratio of the firefly and
Renilla activities and expressed as a percentage of the
control (untreated cells) values.
Preparation of cell lysate and Western
blotting
Cell lysates were prepared in
radioimmunoprecipitation assay buffer, as described previously
(18). Protein content of the
lysates was determined using Pierce Bicinchoninic Acid Assay (cat.
no. 23225; Pierce; Thermo Fisher Scientific, Inc., Rockford, IL,
USA) according to the manufacturer's protocol. Protein samples (100
µg) were separated on a 4–12% NuPAGE
Novex®Bis-Tris gradient mini gel (Thermo Fisher
Scientific, Inc.) and were transferred to a polyvinylidene fluoride
membrane using iBlot (Thermo Fisher Scientific, Inc.). Blots were
blocked for 1 h at room temperature in Tris-buffered saline-0.5%
Tween (TBST) containing 5% dried milk. After blocking, the blots
were washed three times for 5 min in TBST at room temperature.
Blots were subsequently hybridized with the following primary
antibodies diluted in TBST containing 5% nonfat dry milk overnight
at 4°C: Mouse anti-MnSOD (1:100; cat. no. sc-137254; Santa Cruz
Biotechnology, Inc., Dallas, TX) or rabbit anti-shc (1:1,000; cat.
no. 610082; BD Biosciences, San Jose, CA, USA). Subsequently, blots
were washed three times for 5 min in TBST at room temperature and
were then incubated with horseradish peroxidase-labeled anti-mouse
(1:5,000; cat. no. 7076S) or anti-rabbit (1:5,000; cat. no. 7074S)
secondary antibodies (Cell Signaling Technology, Inc., Danvers, MA,
USA) for 45 min at room temperature. Blots were washed a further
three times for 5 min with TBST at room temperature and were
incubated with Pierce Enhanced Chemiluminescence Western Blotting
substrate (Pierce; Thermo Fisher Scientific, Inc.) for 1 min at
room temperature, before being exposed to an X-ray film (Midwest
Scientific, St. Louis, MO, USA). Films were digitized and analyzed
using Un-Scan-It™ Version 6.1 software (Silk Scientific, Orem, UT,
USA). Each blot was stripped with Restore PLUS Western Blot
stripping buffer (cat. no. 46430; Thermo Scientific, Inc.) for 20
min at 37°C and was washed three times for 5 min with TBST at room
temperature. The stripped blots were rehybridized with an
anti-actin antibody (1:20,000; cat. no. MAB1501; EMD Millipore,
Billerica, MA, USA) for 40 min at room temperature, followed by
washing and hybridization with a secondary mouse antibody as
aforementioned.
Statistical analysis
All experiments were performed in triplicate.
Continuous variables are expressed as the mean ± standard
deviation. One-way analysis of variance with Holm-Sidak post-hoc
test was used to evaluate the differences between groups. All
analyses were performed using GraphPad InStat version 3 (GraphPad,
La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
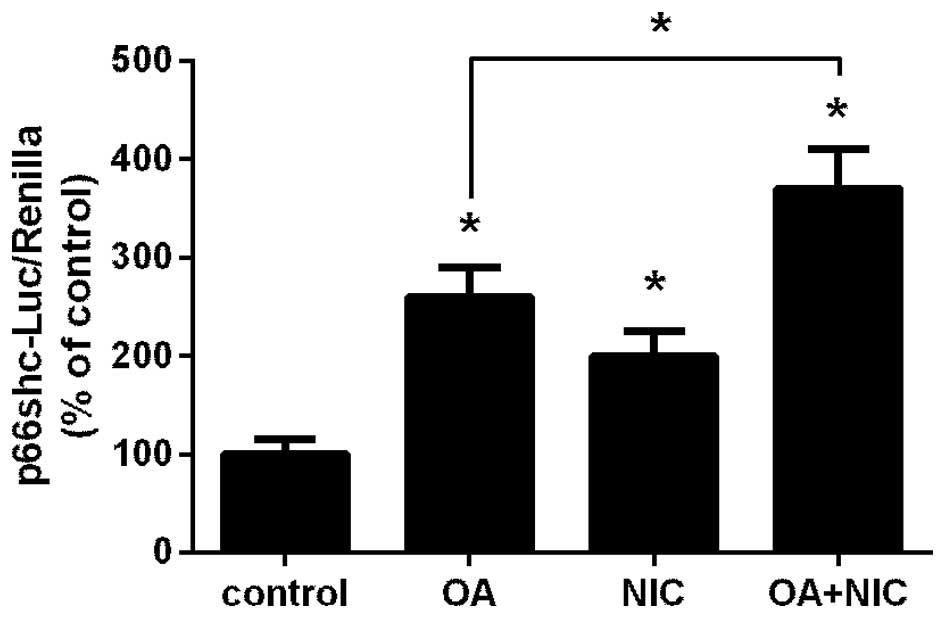
Exposure to NIC augments FFA-dependent
induction of the p66shc promoter
Our previous studies reported that NIC and OA
induced p66shc promoter activity in renal proximal tubule cells
(11,12). Therefore, it is possible that their
combined application is additive. In the present study, NRK52E
cells transfected with a p66shc-promoter luciferase plasmid
(20) together with Renilla
luciferase, were treated with either 100 µM OA, 200
µM NIC, or with a combination of NIC and OA. As shown in
Fig. 1, treatment with OA
(P<0.05) and NIC (P<0.05) significantly increased activity of
the p66shc promoter compared with the control group. In addition,
when both treatments were applied simultaneously, the activity
levels were significantly greater compared with the OA only group
(P<0.05).
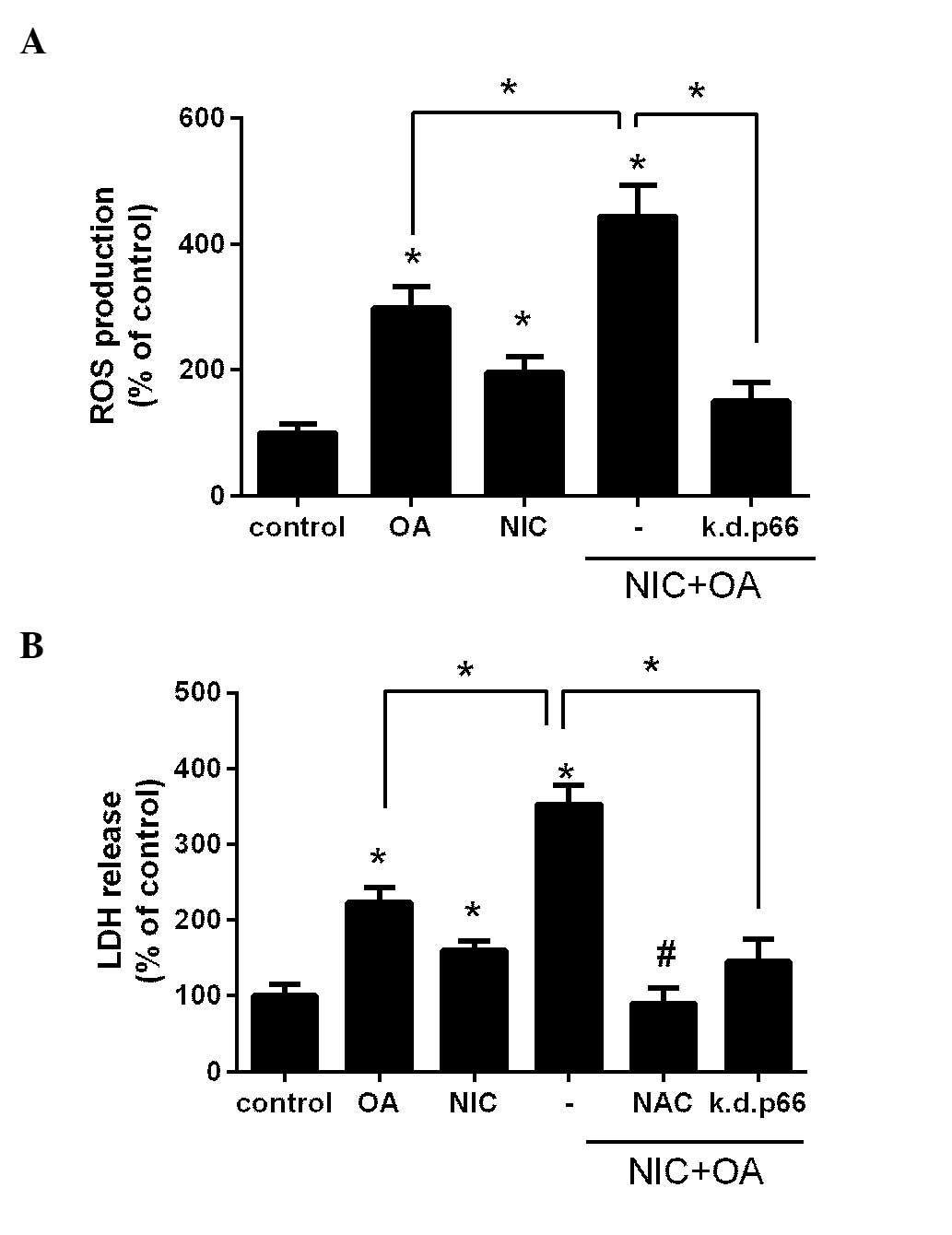
NIC exacerbates FFA-induced ROS
production and consequent injury in a p66shc-dependent manner
Previously, we determined that NIC and OA induced
ROS production and subsequent cell injury via the activation of
p66shc in cultured renal proximal tubule cells (11,12).
In order to determine whether their co-application is additive,
NRK52E cells were treated with either 100 µM OA, 200
µM NIC or with their combination, and intracellular ROS
production was determined. As presented in Fig. 2A treatment with OA and NIC
significantly increased ROS production compared with the control
and OA only groups (P<0.05). In addition, transfection with the
k.d.p66shc plasmid significantly reduced NIC + OA-dependent ROS
production (P<0.05; Fig. 2A).
These findings suggest that the adverse effects of NIC on
OA-mediated ROS release are p66shc-dependent.
The present study also aimed to determine whether an
increase in ROS production by NIC + OA treatment led to increased
cell injury. Cells were treated with OA, NIC or their combination,
and LDH release was determined. As presented in Fig. 2B, treatment with OA and NIC
significantly increased LDH release, and thus cell injury, compared
with the control group (P<0.05). Furthermore, LDH release was
significantly increased in the NIC + OA group compared with the OA
only group (P<0.05). Pretreatment of the cells with the
antioxidant NAC (10 µM) significantly attenuated OA +
NIC-induced cell injury, thus suggesting that NIC + OA-dependent
injury may be mediated through ROS. In addition, transfection with
k.d.p66shc significantly reduced the release of LDH compared with
the NIC + OA treatment group(P<0.05; Fig. 2B), which suggests that the adverse
effects of NIC on OA-mediated cell injury are p66shc-dependent.
NIC attenuates FFA-dependent induction of
MnSOD via p66shc-mediated inactivation of FOXO activity
The present study demonstrated that p66shc acts as a
mediator of the adverse effects of chronic NIC exposure on
OA-dependent lipotoxicity (Fig.
2). In our previous study we reported that NIC mitigates
HFD-dependent induction of MnSOD in the mouse kidney, which also
coincides with increased p66shc expression (10). Therefore, the present study aimed
to investigate whether p66shc is responsible for suppression of
MnSOD. Accordingly, NRK52E cells were transfected with a reporter
luciferase plasmid that harbors the promoter of the MnSOD gene
(21) together with a
Renilla luciferase plasmid, and were treated with either 100
µM OA, 200 µM NIC or with their combination. After 24
h cell luciferase activity was determined. As presented in Fig. 3A, treatment with OA significantly
elevated the MnSOD promoter activity compared with the control
group (P<0.05). However, MnSOD promoter activity was suppressed
in the NIC treatment group. Subsequently, NRK52E cells were
co-transfected with a shp66shc plasmid, to knockdown p66shc
expression, in conjunction with MnSOD-luc reporter/Renilla
luciferase plasmids, and the cells were treated with NIC + OA. As
shown in Fig. 3A, knockdown of
p66shc rescued the OA-dependent induction of MnSOD in the presence
of NIC.
The MnSOD promoter is primarily regulated through
the forkhead FOXO3a transcription factor (23). Previous studies have revealed that
p66shc suppressed MnSOD promoter activity through the inactivation
of FOXO3a in non-renal cells (14,16).
In the present study, NRK52E cells were transfected with a
6xDBE-luc plasmid that contains 6 copies of the canonical FOXO
binding site (22) together with a
Renilla luciferase plasmid, and were subsequently treated
with NIC, OA or with their combination. As presented in Fig. 3B, OA treatment significantly
induced 6xDBE-luciferase activity compared with in the control and
the NIC + OA treatment groups (P<0.05). However, this activity
was suppressed by NIC, which was similar to its effects on the
MnSOD promoter (Fig. 3A). Notably,
k.d.p66shc reduced this suppression (Fig. 3B).
Overexpression of p66shc or suppression
of MnSOD emulates the adverse effects of NIC on OA-mediated
lipotoxicity
The present study indicated that the upregulation of
p66shc and p66shc-mediated suppression of MnSOD may be responsible
for the adverse effects of NIC on OA-mediated lipotoxicity. To
demonstrate this, endogenous MnSOD expression was suppressed by
~70% through transfection of cells with an shMnSOD plasmid
(Fig. 4A). In addition,
transfection with a p66shc expression vector increased the levels
of p66shc by 400%, whereas the levels of the other shc isoforms
(p52shc and p46shc) remained unchanged. Subsequently, these cells
were treated with 100 µM OA, and ROS production and LDH
release were determined. As shown in Fig. 4B, overexpression of p66shc
(OE-p66shc group) significantly increased ROS production compared
with the OA-treated group (P<0.05; Fig. 4B). In addition, knockdown of MnSOD
(k.d.MnSOD group) significantly augmented OA-dependent ROS
production compared with the control group (P<0.05; Fig. 4B). The effects of k.d.MnSOD and
OE-p66shc were similar to the effects of NIC on cell injury and ROS
levels (Fig. 2).
Discussion
Obesity is an epidemic in the United States and
other western countries, and is associated with the increased
incidence of chronic kidney disease in adults and children
(24–26). The potential mechanism underlying
renal lipotoxicity includes FFA-dependent production of ROS
(24), which increases
mitochondrial permeability transition (27) leading to depolarization of the
mitochondria and cell death (28).
OA is a major circulating FFA in obese individuals (29), and our previous study demonstrated
that it increases transcription of the pro-oxidant p66shc gene,
which contributed to increased ROS production and subsequent injury
in cultured renal proximal tubule cells (12).
Although obesity is preventable, many people often
adopt smoking as a practice to lose weight (4,5);
however, they are often oblivious to the fact that the life
expectancy of an obese smoker is 13 years less compared with a
smoker at a normal weight (5).
Adverse effects of smoking on obesity include increased
dyslipidemia (30), diabetes,
insulin resistance, cardiovascular disease (5) and an increased renal risk, the
mechanisms of which remain to be fully elucidated.
Smoking is an independent risk factor that augments
the risk of the increased development and progression of chronic
kidney disease via oxidative stress (31). NIC is a major tobacco alkaloid
(6) that is responsible for the
association between smoking and kidney injury (8). Our previous study demonstrated that
NIC augments oxidative stress via the transcriptional activation of
p66shc, which may be responsible for NIC-mediated ROS production
and consequent cell injury in renal proximal tubule cells (11).
A previous study reported that smoking may
exacerbate the obesity-dependent renal risk (32). Furthermore, NIC in combination with
HFD augments mitochondrial abnormalities (33) and oxidative stress in the liver
(34), which exacerbates the
severity of HFD-induced hepatic lipotoxicity (34). Conversely, to the best of our
knowledge, the impact of NIC on HFD-associated renal lipotoxicity
has not been thoroughly investigated. Our previous study
demonstrated the adverse effects of NIC exposure on HFD-associated
fat deposition and oxidative stress in the kidneys of mice
(10). Our present in vitro
experiments recapitulated this scenario: NIC treatment augmented
OA-dependent ROS production and resulted in increased injury
(Fig. 2). In addition, it was
demonstrated that NIC-induced injury may be mitigated by knockdown
of the p66shc gene (Fig. 2). This
is not surprising, since NIC and OA exert reno-toxicity via the
induction of p66shc expression in cultured renal proximal tubule
cells (11,12). Induction of the p66shc promoter was
increased in cells treated with the combination of NIC and OA
(Fig. 1), which is in agreement
with our previous study in NIC + HFD mice (10). Our previous study demonstrated that
NIC increased p66shc promoter activity via hypomethylation or p53
(11). In addition, it has
previously been reported that the expression levels of p53 in renal
tubular cells are enhanced in obese mice (35) and a genome-wide increase in DNA
methylation has been observed in obese children (36). Whether these pathways act in
conjunction to enhance the effects of p66shc and thus increase
NIC+HFD/FFA-associated renal oxidative stress remains to be
determined.
Increased oxidative stress may be only partially due
to an increase in ROS production; it may also be the result of
impaired antioxidant responses. The present study demonstrated that
NIC increased OA-associated ROS production and suppressed
OA-dependent induction of MnSOD expression in a p66shc-dependent
manner (Fig. 3A); these findings
are in agreement with the results of our previous study in mice fed
a HFD (10). To the best of our
knowledge, this is a novel observation in renal cells and the
molecular mechanism underlying this suppression remains to be
elucidated. Previous studies have demonstrated that p66shc is able
to suppress transcription of MnSOD via inactivation of FOXO3a in
various non-renal cells (14–16).
The results of the present study supported this scenario, since OA
treatment increased the activity of a FOXO reporter (Fig. 3B), which was abolished by NIC.
Notably, knockdown of p66shc expression mitigated the negative
effects of NIC (Fig. 3B), thus
suggesting that p66shc may be responsible for the observed
suppression.
The present study suggested that NIC exerted its
adverse effects by promoting an increase in p66shc-mediated ROS
release and p66shc-induced MnSOD antioxidant suppression. The
overexpression of p66shc and knockdown of MnSOD had similar effects
to NIC treatment on OA-associated renal lipotoxicity (Fig. 4).
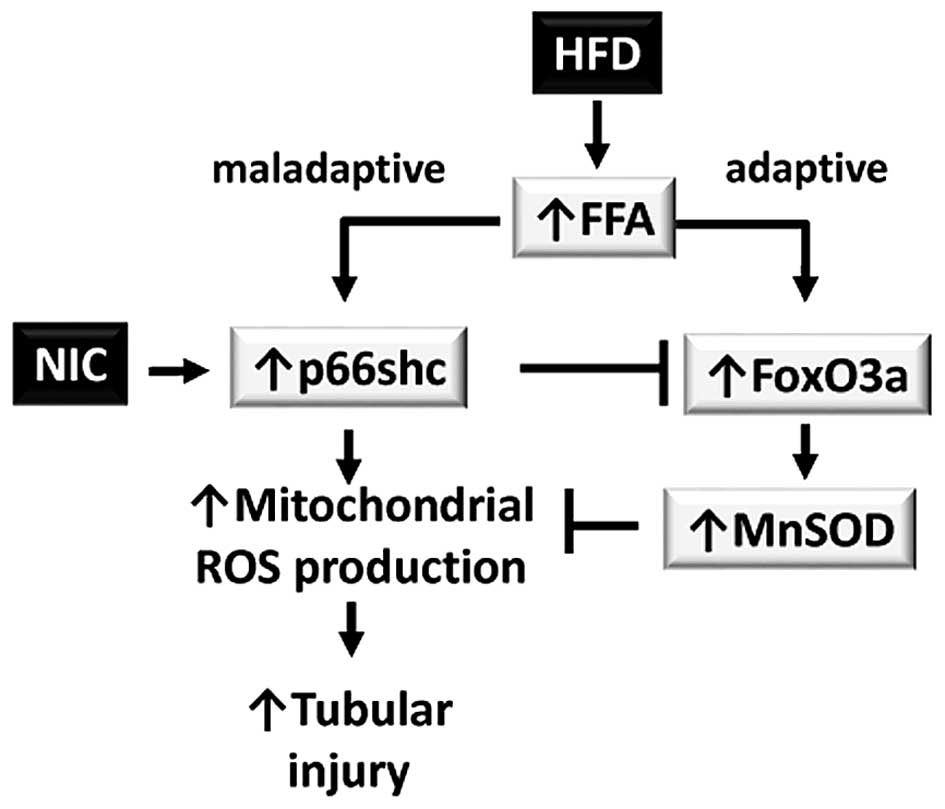
In conclusion, HFD induces maladaptive and adaptive
responses via increased renal FFA deposition. The maladaptive
responses are triggered by transcriptional activation of the p66shc
gene, which increases mitochondrial ROS production and leads to
tubular injury in the kidney. This maladaptive response is
compensated by the adaptive response: FOXO-dependent induction of
the antioxidant gene MnSOD. In the presence of NIC, transcription
of p66shc is increased; therefore, the balance is shifted to a
primarily maladaptive response (Fig.
5).
The overall rate of cigarette smoking in the United
States has been reported to be in decline (37); however, there has been an increase
in the use of alternative NIC delivery products, including
E-cigarettes (38). Perceived as
being a safe alternative to cigarettes (39), the popularity of E-cigarettes is
increasing at an alarming rate (7), which may represent an additional
renal risk to obese smokers. The results of the present study may
offer a therapeutic means to ameliorate the adverse effects of NIC
exposure in obese individuals. Future animal studies are required
to explore therapeutic interventions that aim to modify p66shc
expression.
Acknowledgments
The present study was supported by a grant from the
Department of Pediatrics, University of Mississippi Medical Center
and the Bower Foundation. Many thanks to Dr Irani (Cardiovascular
Institute, University of Pittsburgh Medical Center, Pittsburgh, PA,
USA) for providing the p66shc-promoter luciferase plasmid (20) and to Dr Burgering (Department of
Molecular Cancer Research, University Medical Center Utrecht,
Utrecht, Netherlands) for the MnSOD-promoter (21) and the 6xDBE (22) luciferase plasmids.
References
|
1
|
Schaffer JE: Lipotoxicity: When tissues
overeat. Curr Opin Lipidol. 14:281–287. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto
M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, et al: High
glucose level and free fatty acid stimulate reactive oxygen species
production through protein kinase C-dependent activation of NAD(P)H
oxidase in cultured vascular cells. Diabetes. 49:1939–1945. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weinberg JM: Lipotoxicity. Kidney Int.
70:1560–1566. 2006. View Article : Google Scholar
|
|
4
|
Albanes D, Jones DY, Micozzi MS and
Mattson ME: Associations between smoking and body weight in the US
population: Analysis of NHANES II. Am J Public Health. 77:439–444.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chiolero A, Faeh D, Paccaud F and Cornuz
J: Consequences of smoking for body weight, body fat distribution,
and insulin resistance. Am J Clin Nutr. 87:801–809. 2008.PubMed/NCBI
|
|
6
|
Hukkanen J, Jacob P III and Benowitz NL:
Metabolism and disposition kinetics of nicotine. Pharmacol Rev.
57:79–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schluger NW: The electronic cigarette: A
knight in shining armour or a Trojan horse? Psychiatr Bull (2014).
38:201–203. 2014. View Article : Google Scholar
|
|
8
|
Jaimes EA, Tian RX and Raij L: Nicotine:
The link between cigarette smoking and the progression of renal
injury? Am J Physiol Heart Circ Physiol. 292:H76–H82. 2007.
View Article : Google Scholar
|
|
9
|
Orth SR, Viedt C and Ritz E: Adverse
effects of smoking in the renal patient. Tohoku J Exp Med.
194:1–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Arany I, Hall S, Reed DK, Reed CT and
Dixit M: Nicotine enhances high-fat diet-induced oxidative stress
in the kidney. Nicotine Tob Res. 18:1628–1634. 2016. View Article : Google Scholar
|
|
11
|
Arany I, Clark J, Reed DK and Juncos LA:
Chronic nicotine exposure augments renal oxidative stress and
injury through transcriptional activation of p66shc. Nephrol Dial
Transplant. 28:1417–1425. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arany I, Clark JS, Reed DK, Juncos LA and
Dixit M: Role of p66shc in renal toxicity of oleic acid. Am J
Nephrol. 38:226–232. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Genestra M: Oxyl radicals, redox-sensitive
signalling cascades and antioxidants. Cell Signal. 19:1807–1819.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koch OR, Fusco S, Ranieri SC, Maulucci G,
Palozza P, Larocca LM, Cravero AA, Farre' SM, De Spirito M,
Galeotti T and Pani G: Role of the life span determinant P66(shcA)
in ethanol-induced liver damage. Lab Invest. 88:750–760. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pani G and Galeotti T: Role of MnSOD and
p66shc in mitochondrial response to p53. Antioxid Redox Signal.
15:1715–1727. 2011. View Article : Google Scholar
|
|
16
|
Guo J, Gertsberg Z, Ozgen N and Steinberg
SF: p66Shc links alpha1-adrenergic receptors to a reactive oxygen
species-dependent AKT-FOXO3A phosphorylation pathway in
cardiomyocytes. Circ Res. 104:660–669. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arany I, Faisal A, Clark JS, Vera T,
Baliga R and Nagamine Y: p66SHC-mediated mitochondrial dysfunction
in renal proximal tubule cells during oxidative injury. Am J
Physiol Renal Physiol. 298:F1214–F1221. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arany I, Faisal A, Nagamine Y and
Safirstein RL: p66shc inhibits pro-survival epidermal growth factor
receptor/ERK signaling during severe oxidative stress in mouse
renal proximal tubule cells. J Biol Chem. 283:6110–6117. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Arany I, Grifoni S, Clark JS, Csongradi E,
Maric C and Juncos LA: Chronic nicotine exposure exacerbates acute
renal ischemic injury. Am J Physiol Renal Physiol. 301:F125–F133.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim CS, Jung SB, Naqvi A, Hoffman TA,
DeRicco J, Yamamori T, Cole MP, Jeon BH and Irani K: p53 impairs
endothelium-dependent vasomotor function through transcriptional
upregulation of p66shc. Circ Res. 103:1441–1450. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Essers MA, Weijzen S, de Vries-Smits AM,
Saarloos I, de Ruiter ND, Bos JL and Burgering BM: FOXO
transcription factor activation by oxidative stress mediated by the
small GTPase Ral and JNK. EMBO J. 23:4802–4812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brenkman AB, van den Broek NJ, de Keizer
PL, van Gent DC and Burgering BM: The DNA damage repair protein
Ku70 interacts with FOXO4 to coordinate a conserved cellular stress
response. FASEB J. 24:4271–4280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kops GJ, Dansen TB, Polderman PE, Saarloos
I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH and Burgering
BM: Forkhead transcription factor FOXO3a protects quiescent cells
from oxidative stress. Nature. 419:316–321. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wahba IM and Mak RH: Obesity and
obesity-initiated metabolic syndrome: Mechanistic links to chronic
kidney disease. Clin J Am Soc Nephrol. 2:550–562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gunta SS and Mak RH: Is obesity a risk
factor for chronic kidney disease in children? Pediatr Nephrol.
28:1949–1956. 2013. View Article : Google Scholar
|
|
26
|
Savino A, Pelliccia P, Chiarelli F and
Mohn A: Obesity-related renal injury in childhood. Horm Res
Paediatr. 73:303–311. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hall AM and Unwin RJ: The not so 'mighty
chondrion': Emergence of renal diseases due to mitochondrial
dysfunction. Nephron Physiol. 105:p1–p10. 2007. View Article : Google Scholar
|
|
28
|
Lemasters JJ, Nieminen AL, Qian T, Trost
LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA,
Brenner DA and Herman B: The mitochondrial permeability transition
in cell death: A common mechanism in necrosis, apoptosis and
autophagy. Biochim Biophys Acta. 1366:177–196. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gray DS, Takahashi M, Bauer M and Bray GA:
Changes in individual plasma free fatty acids in obese females
during fasting and refeeding. Int J Obes. 15:163–168.
1991.PubMed/NCBI
|
|
30
|
Craig WY, Palomaki GE and Haddow JE:
Cigarette smoking and serum lipid and lipoprotein concentrations:
An analysis of published data. BMJ. 298:784–788. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Orth SR and Hallan SI: Smoking: A risk
factor for progression of chronic kidney disease and for
cardiovascular morbidity and mortality in renal patients-absence of
evidence or evidence of absence? Clin J Am Soc Nephrol. 3:226–236.
2008. View Article : Google Scholar
|
|
32
|
Moyad MA: Obesity, interrelated
mechanisms, and exposures and kidney cancer. Semin Urol Oncol.
19:270–279. 2001.
|
|
33
|
Sinha-Hikim I, Friedman TC, Shin CS, Lee
D, Ivey R and Sinha-Hikim AP: Nicotine in combination with a
high-fat diet causes intramyocellular mitochondrial abnormalities
in male mice. Endocrinology. 153:865–872. 2014. View Article : Google Scholar
|
|
34
|
Friedman TC, Sinha-Hikim I, Parveen M,
Najjar SM, Liu Y, Mangubat M, Shin CS, Lyzlov A, Ivey R, Shaheen M,
et al: Additive effects of nicotine and high-fat diet on hepatic
steatosis in male mice. Endocrinology. 153:5809–5820. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brezniceanu ML, Liu F, Wei CC, Chénier I,
Godin N, Zhang SL, Filep JG, Ingelfinger JR and Chan JS:
Attenuation of interstitial fibrosis and tubular apoptosis in db/db
transgenic mice overexpressing catalase in renal proximal tubular
cells. Diabetes. 57:451–459. 2008. View Article : Google Scholar
|
|
36
|
Huang RC, Garratt ES, Pan H, Wu Y, Davis
EA, Barton SJ, Burdge GC, Godfrey KM, Holbrook JD and Lillycrop KA:
Genome-wide methylation analysis identifies differentially
methylated CpG loci associated with severe obesity in childhood.
Epigenetics. 10:995–1005. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lauterstein D, Hoshino R, Gordon T,
Watkins BX, Weitzman M and Zelikoff J: The changing face of tobacco
use among United States youth. Curr Drug Abuse Rev. 7:29–43. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grana R, Benowitz N and Glantz SA:
E-cigarettes: A scientific review. Circulation. 129:1972–1986.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baeza-Loya S, Viswanath H, Carter A,
Molfese DL, Velasquez KM, Baldwin PR, Thompson-Lake DG, Sharp C,
Fowler JC, De La Garza R II and Salas R: Perceptions about
e-cigarette safety may lead to e-smoking during pregnancy. Bull
Menninger Clin. 78:243–252. 2014. View Article : Google Scholar : PubMed/NCBI
|