Introduction
Renal cell carcinoma (RCC) is a public health
problem worldwide, and is responsible for significant
cancer-associated mortalities, accounting for more than 3% of all
malignancies. Patients with RCC have a median survival time of 13
months. As the predominant form (>80%) of RCC, clear cell RCC
(CCRCC) is highly aggressive and unresponsive to radiation or
chemotherapy (1). The combination
of surgery with radiotherapy or chemotherapy is the most effective
treatment option for the majority of patients; however, patients
with CCRCC often suffer recurring tumors as distant metastases,
with only 4–6% of them responding to chemotherapy. One of the most
important current treatment strategies is the administration of
interleukin-2 (IL-2). However, high doses of IL-2 exhibit a low
response rate with marked toxicity to patients (1). Although various other drugs,
including the vascular endothelial growth factor receptor-2,
platelet-derived growth factor and the receptor-β inhibitors,
sorafenib (Nexavar) and sunitinib (Sutent), have been used for
clinical trials, they did not prove to be highly effective in
inhibiting the growth of metastatic CCRCC cells (2).
Inactivation of the von Hippel-Lindau tumor
suppressor (VHL; E3 ubiquitin ligase gene) is associated with RCC
(3,4), particularly in the case of CCRCC.
Sporadic CCRCC may arise due to the biallelic inactivation of the
VHL protein, and is characterized by somatic mutations or
hypermethylation of the VHL region (5,6). VHL
encodes two protein isotypes with molecular masses of 30 and 19
kDa. They have different functions due to their subcellular
localization (7). Although
hypoxia-inducible factor (HIF)-α is a confirmed substrate of VHL,
the larger (30 kDa) isotype of VHL was reported to bind to P53
through its α domain (155–213 amino acids) (6). The association of VHL and P53
stabilized P53 by suppressing mouse double minute 2 homolog
(MDM2)-induced ubiquitination and the nuclear export of P53
(8). This has also been identified
to increase the transcriptional activity of P53 and P53-mediated
cell cycle arrest and apoptosis (8).
The P53 protein, encoded by tumor protein p53
(TP53), controls multiple cellular functions, including cell
proliferation, DNA repair, senescence and apoptosis (9). Dysfunction of, and mutations in, the
P53 signaling pathway were also reported to contribute to the
resistance of tumors to chemotherapeutic agents through a failure
of transcriptional regulation of its target genes, such as
cyclin-dependent inhibitor 1A (also termed P21), Bcl2-associated X
protein (Bax) and B-cell CCL/lymphoma 2 (10), suggesting that the efficacy of
chemotherapy often depends on P53-mediated cell cycle arrest and
apoptosis. Thus, P53 mutations are associated with chemoresistance
and, if present, often are predictors of an unfavorable prognosis
for patients (11). Although
>50% of P53 genes are mutated in tumors, it is notable that P53
mutations are infrequently detected in CCRCC (12). The predominant research focus, to
the best of our knowledge, has been to elucidate the mechanism
underlying the development of resistance in CCRCC. CCRCC cells
lacking VHL are resistant to tumor necrosis factor (TNF) and
receptor-mediated cell death due to the upregulation of nuclear
factor κB (NF-κB) (13). However,
studies on the role of P53 in the development of chemoresistance in
RCC or CCRCC have not been widely reported, and the mechanism
underlying the involvement of P53 in the development of
chemoresistance in RCC or CCRCC has yet to be fully elucidated.
Notably, CCRCC with functional P53 is commonly insensitive to
chemotherapy, thus indicating that P53 dysfunction is not the only
mechanism contributing to the progression of CCRCC. In the present
study, it was hypothesized that the stabilization of P53 by VHL,
which leads to an inhibition of its ubiquitination, may be the
potential mechanism to account for how the elimination of
functional P53 causes chemosensitivity.
Although there is no general consensus, RCC cells
lacking VHL are often resistant to chemotherapy (13). The most common form (75%) of kidney
cancer is CCRCC, which is resistant to radiation and chemotherapy
when VHL is mutated (1) The
current study determined that the proper functioning of VHL and P53
is required for chemosensitivity in RCC and CCRCC. VHL stabilized
P53 by inhibiting its ubiquitination following chemotherapy, and
led to the upregulation of the target genes of P53, P21 and Bax.
Consequently, upregulation of P21 led to cell cycle arrest at the
G2 phase and completely inhibited cell proliferation, whereas the
upregulated level of Bax induced apoptosis. These findings provided
the potential mechanistic link between VHL and P53-dependent
chemoresistance in CCRCC.
Material and methods
Cell cultures
The cell lines used in the current study, 786-O,
ACHN and MZ1257, were purchased from the American Type Culture
Collection (Manassas, VA, USA). 786-O and ACHN cells were cultured
in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.). MZ1257
cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS. Cells were cultured in a
humidified atmosphere of 5% CO2 at 37°C.
Overexpression of VHL or P53
The VHL expression plasmid, pcDNA3-VHL, and the P53
expression plasmid, pcDNA3-P53, were kept in our laboratory.
Briefly, the coding sequences of VHL or P53 was amplified and
inserted behind the cytomegalovirus promoter of pcDNA3 expression
vector, respectively. The cells were transfected with 0.8 µg
of the desired plasmid using Lipofectamine 2000 (Thermo Fisher
Scientific, Inc.). After 24 h, G418 (600 µg/ml for 786-O;
450 µg/ml for MZ1257; Thermo Fisher Scientific, Inc.) was
added for 2 weeks in order to select for cells that had taken up
the plasmids. Colonies were individually selected, and subsequently
were analyzed by semiquantitative western blot analysis.
RNA interference
Plasmid-based short hairpin (sh)RNAs were
constructed, with target sequences 5′-GAGAACTGGGACGAGGCCG-3′ (for
VHL shRNA) (14) and
5′-GACTCCAGTGGTAATCTAC-3′ (for P53 shRNA) (15). The shRNAs were cloned into a
pcDNA-based plasmid downstream of the U6 promoter. Cells which
stably expressed the plasmid were obtained as described above.
Functional agonist assay
The cell lines ACHN (containing wild-type VHL),
786-O (VHL null) and MZ1257 (VHL null) were selected for ADM and
sunitinib treatment. The IC50 values (i.e. the
concentration required to give half-maximal inhibition) of
adriamycin (ADM) and Sunitinib for 786-O, MZ1257 and ACHN cells
were determined using a Cell Counting Kit-8 (Shanghai Shenggong
Biology Engineering Technology Service, Ltd., Shanghai, China) to
detect cells with metabolic activity. The absorbance at 450 nm was
quantified using a microplate reader (Thermo Fisher Scientific,
Inc.) following treatment with the drugs. Cells (2×104)
suspended in DMEM supplemented with 10% FBS were seeded into 6-well
plates and incubated overnight at 37°C. Cells were treated with ADM
(1, 2, 5, 10, 20 and 50 mg/l) or sunitinib (1, 2.5, 5, 7.5, 10, 15
and 20 µM) were added in the medium for 24 h treatment.
Flow cytometric analysis
Following treatment with the drugs, the cells were
trypsinized (trypsin; Thermo Fisher Scientific, Inc.) and harvested
the cells were trypsinized and harvested by centrifugation using an
Eppendorf 5415D (Eppendorf, Hamburg, Germany) at 4°C, 1000 × g.
Then cells were fixed in 70% ethanol (Shanghai Shenggong Biology
Engineering Technology Service, Ltd.) pre-cooled to 4°C for 12–24 h
pre-cooled to 4°C for 12–24 h. Subsequently, the cells were washed
three times using ice-cold phosphate-buffered saline for 5 min.
Fixed cells were subjected to propidium iodide (PI; Sigma-Aldrich,
St. Louis, MO, USA)/ribonuclease (RNase; Shanghai Shenggong Biology
Engineering Technology Service, Ltd.) staining. Flow cytometric
analysis was determined using a flow cytometer (Navios; Beckman
Coulter, Inc., Brea, CA, USA). Briefly, cells (1×106)
were resuspended in 0.5 ml binding buffer (Abcam, Cambridge, UK)
containing 5 µl annexin V and 5 µl PI, and incubated
at room temperature for 30 min in the dark. The apoptotic rate was
detected by flow cytometry. For the flow cytometric analysis of
apoptosis, the apoptotic rate was assessed using an annexin
V-fluorescein isothiocyanate apoptosis detection kit (Beyotime
Institute of Biotechnology, Haimen, China), according to the
manufacturer's protocol.
Western blot analysis
The total protein was extracted using RIPA lysis
buffer (Thermo Fisher Scientific, Inc.) by adding Halt Protease
Inhibitor Cocktail (Thermo Fisher Scientific, Inc.) to prevent
protein degradation. The Pierce BCA Protein Assay kit (Thermo
Fisher Scientific, Inc.) was used to determine the protein
concentration that was extracted. For semiquantitative analysis,
equivalent quantities of protein were resolved and mixed with 5X
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(SDS-PAGE) loading buffer (Shanghai Shenggong Biology Engineering
Technology Service, Ltd.) containing 10% SDS, and transferred onto
a polyvinylidene fluoride membrane (Merck Millipore, Darmstadt,
Germany) at 110 V for 1.5 h. Subsequently, the blotted membranes
were blocked with 5% bovine serum albumin in Tris-buffered saline
and then incubated with primary antibodies as follows: Monoclonal
mouse anti-human VHL antibody (cat no. ab140989;1:1,000; monoclonal
mouse anti-human P53 antibody, (cat no. ab28; 1:2,000; and
monoclonal mouse anti-human β-actin antibody, (cat no. ab182951;
1:5,000), all from Abcam, Cambridge, UK). β-actin was detected as
the housekeeping gene. The step was followed by incubation with the
relevant horseradish peroxidase-conjugated secondary antibody,
including: Anti-VHL, cat no. ab140989, 1:1,000; anti-p53 antibody,
cat no. ab28, 1:2,000; and anti-gamma actin antibody, 1:2,000; cat
no. ab182951, all rabbit anti-mouse IgG Ab; Abcam. The signal was
detected using an enhanced chemiluminescence detection system
(Pierce ECL Western Blotting Substrate; Thermo Fisher Scientific,
Inc.).
Statistical analysis
Quantitative results are expressed as the mean ±
standard deviation. Differences between groups were analyzed using
one-way analysis of variance, followed by Bonferroni post hoc
analysis. Statistical analysis was performed using SPSS software
(version 19.0; SPSS, Inc., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
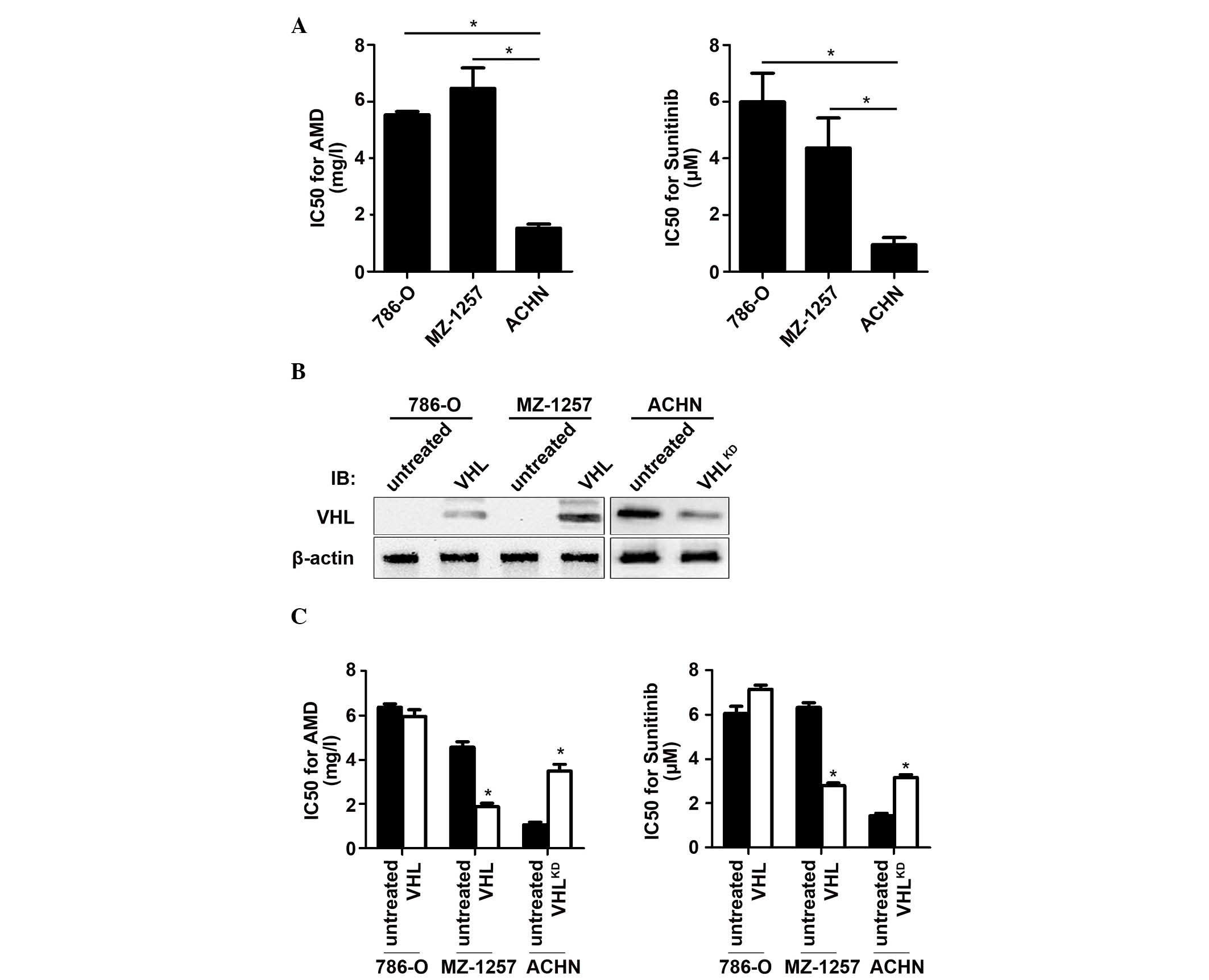
Expression of VHL contributes to
chemosensitivity in CCRCC cells
Therefore, the aim of the current study was to
investigate the importance of VHL in the development of
chemoresistance in CCRCC. The functional agonist assay, which was
used to assess the half-maximal inhibition of cell proliferation
(IC50 values), indicated that, compared with 786-O and
MZ1257, which were VHL null, the ACHN cell line was sensitive to
ADM and sunitinib (Fig. 1A). To
confirm that VHL contributes to chemosensitivity, VHL was
introduced into the 786-O (786-O-VHL) or the MZ1257 (MZ1257-VHL)
cells, or was knocked down by introducing shRNA into the ACHN
(ACHN-VHLKD) cells (Fig.
1B). These results indicated that the expression of VHL led to
cell chemosensitivity in MZ1257-VHL, and that suppression of VHL
reduced the chemosensitivity in ACHN-VHLKD cells
(Fig. 1C).
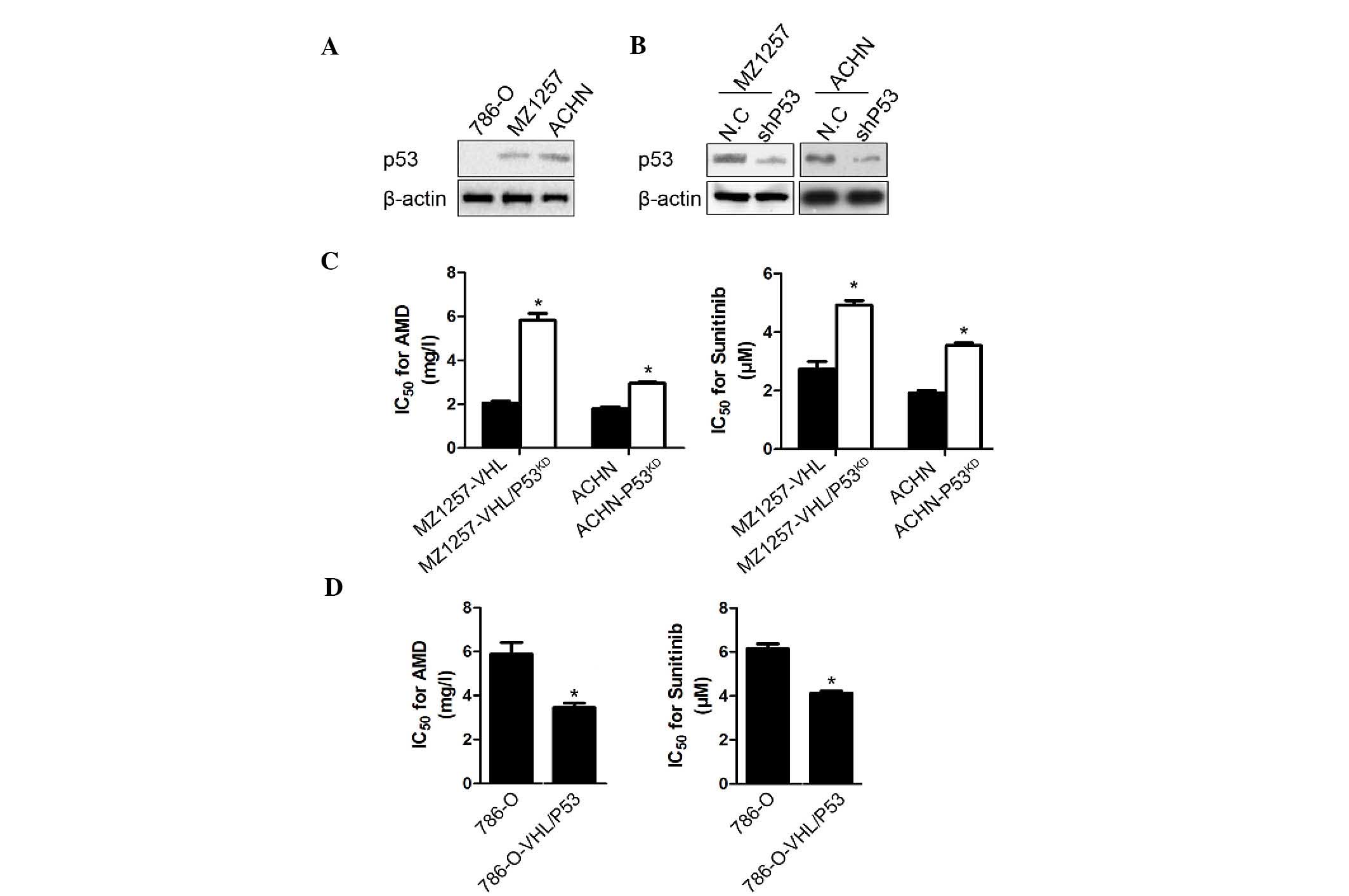
Presence of VHL and P53 is required for
chemosensitivity in RCC cells
The introduction of VHL into the MZ1257 cells led to
a greater sensitivity to ADM and sunitinib treatment; however, the
786-O-VHL cells exhibited no detectable change on treatment with
the drugs (Fig. 1C). The main
difference between the 786-O and MZ1257 cell lines was that 786-O
is P53 null, whereas MZ-1257 contains wild-type P53, which is shown
in Fig. 2A by the western blot
analysis. Therefore, the present study focused on the expression of
P53 and its contribution to CCRCC chemosensitivity. In the ACHN and
MZ1257-VHL cells, when P53 was knocked down by the shRNA target to
P53 mRNA, their chemosensitivity to ADM and sunitinib decreased
markedly (Fig. 2B). Compared with
the MZ1257-VHL cells, the chemosensitivity of the
MZ1257-VHL/P53KD cells to ADM and sunitinib decreased
and became similar to that of MZ1257, which indicated the
importance of P53 in chemosensitivity (Fig. 2C). Co-expression of VHL and P53 in
the 786-O cells led to an even greater sensitivity to chemotherapy
(Fig. 2D).
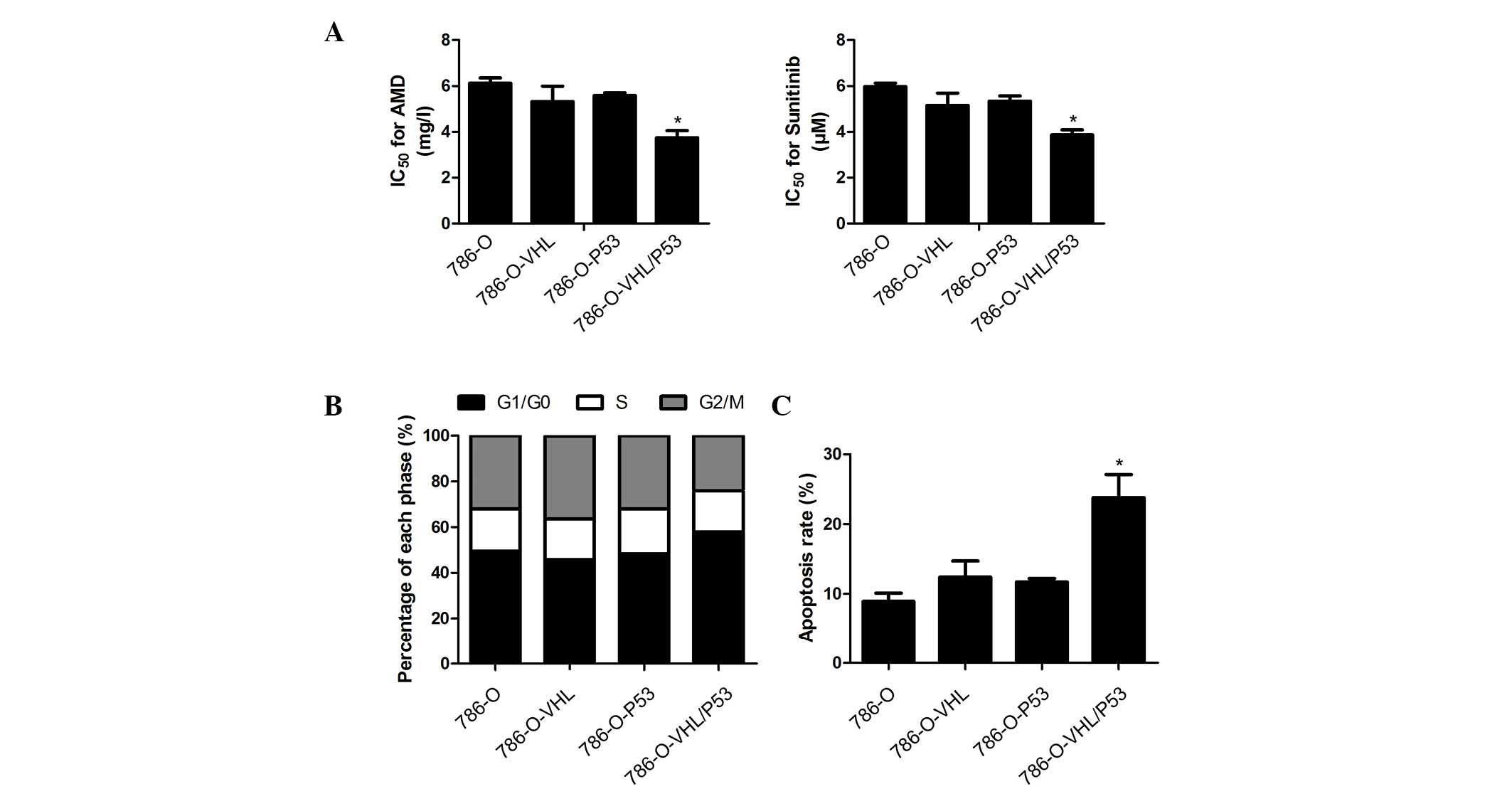
Presence of VHL and P53 enhances
apoptosis following treatment with drugs in CCRCC
In order to confirm the importance of VHL and P53 in
CCRCC chemosensitivity, the 786-O, 786-O-VHL, 786-O-P53 and
786-O-VHL/P53 cell lines were employed in the further analyses. The
cell survival time and chemosensitivity to sunitinib are presented
in Fig. 3A, which shows that,
compared with the original cells, cells co-expressing VHL and P53
were notably more sensitive to sunitinib. However, cells expressing
VHL or P53 alone did not exhibit an increased chemosensitivity to a
similar extent, thereby indicating the importance of co-expression
of VHL and P53 in enhancing chemosensitivity in CCRCC. As p53 is an
established cell cycle regulator, the cell cycle progression was
investigated using flow cytometric analysis. The cell cycles of the
786-O-VHL/P53 and 786-O-P53 cells, compared with those of 786-O and
786-O-VHL, were revealed to be arrested at G0/G1 prior to sunitinib
treatment. Additionally, following sunitinib treatment, the G0/G1
phase in the 786-O-VHL/P53 cells was significantly higher compared
with 786-O-P53, demonstrating the synergistic effect of VHL and P53
in CCRCC (Fig. 3B). The apoptotic
rate was further examined 48 h following treatment with the drugs.
The flow cytometric data revealed that, following sunitinib
treatment, the apoptotic rate of 786-O-VHL/P53 was 24.1±3.4%, which
was markedly higher compared with that of any of the other cell
groups (Fig. 3C).
Discussion
In the kidney, the loss of functional tumor
suppressor gene VHL often leads to genetic or epigenetic
dysfunction, and is an early event that promotes tumorigenesis,
leading to CCRCC. As the most common form (75%) of renal cancer,
CCRCC is typically aggressive and chemoresistant (1). VHL is important in sensitizing RCC
and CCRCC to chemotherapeutic agents. Qi and Ohh (13) reported that CCRCC cells that are
devoid of functional VHL and are resistant to TNF-α-mediated
cytotoxicity (RC3 cells) restored their sensitivity to TNF-α
cytotoxicity following VHL reconstitution. An et al
(16) also determined that the
expression of VHL sensitizes RCC cells to bortezomib by reducing
constitutive NF-κB activity.
In the majority of cancers, the efficacy of
chemotherapy is dependent on a successful execution of p53-mediated
apoptosis induced by the chemotherapeutic agent. Accordingly, it is
reported that, in CCRCC, VHL and P53 act in synergy to promote
sensitization to chemotherapeutic agents. In CCRCC, the
overexpression of HIF-2α, which is induced by the loss of VHL,
leads to MDM2-mediated suppression of P53 and promotes
chemoresistance to sunitinib (17). Functional P53 may be restored by
downregulating MDM2, HIF2α or reconstituting VHL, thereby reversing
chemoresistance. Although >50% of P53 genes are mutated in
cancer cells, P53 mutations are infrequently detected in CCRCC
(12,18). It may have been anticipated that
this infrequent mutation rate of P53 would be beneficial in the
chemosensitivity of CCRCC; however, this has proven not to be the
case. It is possible that both VHL and P53 are required for CCRCC
to be sensitive to chemotherapeutic agents.
The present study determined that VHL and P53 are
required for CCRCC to be chemosensitive to chemotherapeutic agents.
First, CCRCC cells that express wild-type VHL (ACHN) are more
sensitive to chemotherapeutic agents, including ADM and sunitinib,
when compared with VHL-deficient (786-O) or P53-deficient (MZ1257)
CCRCC cells. Secondly, reconstitution of VHL in MZ1257 cells
promoted resistance to the chemotherapeutic agent, although not in
786-O cells, suggesting the necessity of both VHL and P53. Thirdly,
786-O cells co-expressing VHL and P53 were more sensitive to
chemotherapeutic agents when compared with the 786-O, 786-O-VHL or
786-O-P53 cells, indicating the synergistic effect of VHL and
P53.
In conclusion, the current study determined that
co-expression of VHL and P53 sensitizes cells to ADM and sunitinib.
This may have relevance for the clinical management of CCRCC.
Specifically, the present findings suggest that VHL and P53 may be
used as molecular markers to identify patients with CCRCC who would
benefit from ADM or sunitinib therapy. The present study provides
important implications in the diagnosis and treatment of CCRCC.
Acknowledgments
The authors would like to thank Dr Huimin Shi for
suggestions throughout the execution of the present study, and for
editing the English.
References
|
1
|
Cohen HT and McGovern FJ: Renal-cell
carcinoma. N Engl J Med. 353:2477–2490. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brugarolas J: Renal-cell carcinoma -
molecular pathways and therapies. N Engl J Med. 356:185–187. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaelin WG Jr: Molecular basis of the VHL
hereditary cancer syndrome. Nat Rev Cancer. 2:673–682. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernardi R, Scaglioni PP, Bergmann S, Horn
HF, Vousden KH and Pandolfi PP: PML regulates p53 stability by
sequestering Mdm2 to the nucleolus. Nat Cell Biol. 6:665–672. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luu VD, Fischer B, von Teichman A, Boysen
G, Mertzk K, Zimmermann P, Moch H and Schraml P: Von-Hippel Lindau
gene mutation types. Association of gene expression signatures in
clear cell renal cell carcinoma. Pathologe. S2:303–307. 2008.In
German. View Article : Google Scholar
|
|
6
|
Rechsteiner MP, von Teichman A, Nowicka A,
Sulser T, Schraml P and Moch H: VHL gene mutations and their
effects on hypoxia inducible factor HIFα: Identification of
potential driver and passenger mutations. Cancer Res. 71:5500–5511.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iliopoulos O, Ohh M and Kaelin WG Jr:
pVHL19 is a biologically active product of the von Hippel-Lindau
gene arising from internal translation initiation. Proc Natl Acad
Sci USA. 95:11661–11666. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roe JS, Kim H, Lee SM, Kim ST, Cho EJ and
Youn HD: p53 stabilization and transactivation by a von
Hippel-Lindau protein. Mol Cell. 22:395–405. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Murray-Zmijewski F, Slee EA and Lu X: A
complex barcode underlies the heterogeneous response of p53 to
stress. Nat Rev Mol Cell Biol. 9:702–712. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gasco M and Crook T: p53 family members
and chemoresistance in cancer: What we know and what we need to
know. Drug Resist Updat. 6:323–328. 2003. View Article : Google Scholar
|
|
11
|
Wallace-Brodeur RR and Lowe SW: Clinical
implications of p53 mutations. Cell Mol Life Sci. 55:64–75. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tomasino RM, Morello V, Tralongo V, Nagar
C, Nuara R, Daniele E, Curti M and Orestano F: p53 expression in
human renal cell carcinoma: An immunohistochemical study and a
literature outline of the cytogenetic characterization.
Pathologica. 86:227–233. 1994.PubMed/NCBI
|
|
13
|
Qi H and Ohh M: The von Hippel-Lindau
tumor suppressor protein sensitizes renal cell carcinoma cells to
tumor necrosis factor-induced cytotoxicity by suppressing the
nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res.
63:7076–7080. 2003.PubMed/NCBI
|
|
14
|
Kondo K, Kim WY, Lechpammer M and Kaelin
WG Jr: Inhibition of HIF2alpha is sufficient to suppress
pVHL-defective tumor growth. PLoS Biol. 1:E832003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jung DJ, Jin DH, Hong SW, Kim JE, Shin JS,
Kim DJ, Cho BJ, Hwang YI, Kang JS and Lee WJ: Foxq3 expression in
p53-dependent DNA damage responses. J Biol Chem. 285:7995–8002.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
An J, Fisher M and Rettig MB: VHL
expression in renal cell carcinoma sensitizes to bortezomib
(PS-341) through an NF-kappaB-dependent mechanism. Oncogene.
24:1563–1570. 2005. View Article : Google Scholar
|
|
17
|
Roberts AM, Watson IR, Evans AJ, Foster
DA, Irwin MS and Ohh M: Suppression of hypoxia-inducible factor
2alpha restores p53 activity via Hdm2 and reverses chemoresistance
of renal carcinoma cells. Cancer Res. 69:9056–9064. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vasavada SP, Novick AC and Williams BR:
P53, bcl-2, and Bax expression in renal cell carcinoma. Urology.
51:1057–1061. 1998. View Article : Google Scholar : PubMed/NCBI
|