Introduction
Cardiovascular disease, particularly, coronary
artery disease, is a leading contributor to mortality rates
worldwide. Compelling evidence suggests that endothelial
dysfunction, which results from the reduction of nitric oxide (NO)
and the impairment of NO bioavailability, is a key early event in
the pathogenesis of atherosclerosis and an important predictor of
cardiovascular disease (1–4).
Insulin resistance, the early stage of type 2
diabetes, is not only accompanied by glucose and lipid metabolism
disorder, but is also associated with endothelial dysfunction
(5). A number of studies have
suggested that insulin resistance is one of the independent risk
factors for the progression of cardiovascular diseases (6,7).
The reciprocal association between insulin
resistance and endothelial dysfunction in the development of
diabetes and cardiovascular disease has been suggested in animal
studies (8,9). Notably, studies have suggested that
the impairment of insulin signaling results in the reduced
production of NO in an animal model (10,11).
In addition, animal and clinical studies have demonstrated that
endothelial cells exhibit dysfunction in response to high glucose
concentrations (12,13), or prolonged modest hyperinsulinemia
(14,15). However, in vivo, several
complicated factors, including individual differences in
experimental and insulin receptor sensitivity, affect endothelial
dysfunction; thus a reproducible and highly stable endothelial cell
model of endothelium-specific insulin resistance is essential to
examine the specific insulin signaling associated with endothelial
dysfunction.
Atorvastatin, a vessel protective drug, have been
confirmed to reduce cardiovascular events in patients by protecting
endothelial function, in addition to decreasing levels of
cholesterol (16). This drug has
pleiotropic effects, including the upregulation and activation of
endothelial NO synthase (eNOS), which in turn causes an increase in
the production of NO, and reduces oxidative stress and vascular
inflammation (17,18). Phosphatidylinositol 3-kinase (PI3K)
is one of the most well-characterized downstream effectors of
insulin receptor substrate-1 (IRS-1) proteins (19). In vivo studies have
suggested that atorvastatin improves endothelial function via the
phosphatidylinositol 3-kinase (PI3K)/Akt/eNOS pathway (20–22).
In the present study, an endothelial cell model of
endothelium-specific insulin resistance was established, which was
induced by high concentrations of glucose in combination with high
concentrations of insulin. Subsequently, the cell model was treated
with atorvastatin to evaluate its effect on insulin
resistance-associated endothelial dysfunction and to identify the
potential pathway responsible for its action.
Materials and methods
Cell culture
Human umbilical vein endothelial cells (HUVECs)
(23), provided by the Department
of Pathology, Jilin University (Jilin, China), were purchases from
Cobioer Biosciences (Nanjing, China). The cells were maintained at
37°C in humidified air (5% CO2) and cultured in
serum-free Dulbecco's modified Eagle's medium (DMEM; GE Healthcare
Life Science HyClone Laboratories, Logan, UT, USA) with 5 mM
D-glucose (GE Healthcare Life Science HyClone Laboratories), 4 mM
L-glutamine, and 20% fetal bovine serum (GE Healthcare Life Science
HyClone Laboratories).
Treatment of HUVECs with high glucose and
high insulin concentrations to impair insulin signaling
HUVECs (105/ml) were incubated in culture
medium containing 5, 15 or 30 mM D-glucose. The cells were
maintained at 37°C in humidified air (5% CO2) with or
without 10−5 M insulin for 24 h. The medium was then
discarded, the cells were washed twice with phosphate-buffered
saline (PBS), and were then incubated for 6 h following the
addition of 10−5 M insulin at 37°C with 5%
CO2.
Treatment with atorvastatin
HUVECs (106/ml) cultured in normal
culture medium (Nor vec) and HUVECs pretreated with 30 mM glucose
and 10−5 M insulin for 24 h at 37°C with 5%
CO2, defined as insulin-resistant HUVECs (IR vec), were
treated with varying concentrations of atorvastatin (0,
10−6, 10−5 and 10−4 M) for 24 h or
were treated with 10−4 M atorvastatin for 0, 3, 6, 9,
12, 15, 18, 21 and 24 h at 37°C with 5% CO2.
Treatment with LY29004, an inhibitor of
PI3K
The Nor vec and IR vec cells (106/ml)
were treated with PBS (control), 10−4 M atorvastatin
only, 25 µM LY29004 (Merck Millipore, Darmstadt, Germany)
alone or with a combination of 10−4 M atorvastatin and
25 µM LY29004 for 24 h at 37°C with 5% CO2.
Western blot analysis
The HUVECs were pretreated, as described above, in
10 cm cell dishes. Cell lysates were prepared by washing twice in
cold PBS followed by lysis buffer, containing 50 mM Tris-HCl (pH
7.4), 10 mM EDTA, 5 mM EGTA, 0.5% NP40, 1% Triton X-100 and
protease inhibitor (Roche, Mannheim, Germany) and sonication. The
cell lysate was centrifuged at 14,000 × g for 10 min at 4°C, and
the supernatant was discarded. Lysis buffer (150–200 µl) was
added to each Eppendorf tube. Protein concentration was measured
using a Pierce BCA protein assay kit (Thermo Fisher Scientific,
Inc. Waltham, MA, USA). Protein samples (40–60 µg) were
mixed with SDS-PAGE loading dye, boiled and loaded onto an SDS-PAGE
gel (4–20%; Roche). Following transfer of the proteins onto PVDF
membranes (Roche), the proteins were blotted with anti-IR (cat. no.
sc-57344), anti-phosphorylated (phosphor)-IR (tyrosine; cat. no.
sc-17200), anti-IRS-1 (cat. no. SC-8038) and anti-phosphor-IRS-1
(tyrosine; cat. no. sc-17194) antibodies from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), and anti-phosphor-eNOS
(serine1177) and total eNOS antibody (cat. no. orb6756) from BD
Transduction Laboratories (San Diego, CA, USA) in Tris-buffered
saline with Tween, containing 50 mM Tris (pH 7.5), 0.15 M NaCl and
0.05% Tween-20, with 5% fat free milk for 24 h at 4°C. Following
incubation with antibodies, the membranes were washed and then
reacted with horseradish-peroxidase-conjugated IgG (Santa Cruz
Biotechnology, Inc.) for 1 h at 4°C. The membranes were then
treated with ECL reagent (GE Healthcare, Piscataway, NJ, USA) and
exposed to X-ray film. β-actin was used as the protein loading
control. Each experiment contained three replicates and was
repeated three times.
Analysis of total NO concentrations
Total NO concentrations were determined using a
nitrate reduction assay, in which the summation of
NO2− and NO3− (24) were measured using a commercially
available NO kit (Nanjing Jiancheng Biological Products Co., Ltd.,
Nianjing, China), according to the manufacturer protocols. This
assay was used to quantify the dose effects, as measured by the
absorbance at 550 nm. The following equation was used: NO
(µM) = (A − A0) / (As − A0) * Cs * P; where A=sample
absorbance, A0=control absorbance As=standard absorbance,
Cs=standard concentration (100 µM), P=dilution factor. Each
experiment contained six replicates and was repeated three
times.
Reverse transcription-quantitative
polymerase chain reaction analysis (RT-qPCR) of the mRNA levels of
ET-1
Following the 24 h incubation period, total RNA was
extracted from the HUVECs using TRIzol reagent (Sigma-Aldrich, St.
Louis, MO, USA), according to the manufacturer's protocol. Total
RNA (2 µg) was subjected to random-primed reverse
transcription using SuperScript-2 reverse transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.). qPCR was performed in
triplicate using an Applied Biosystem 7900 HT system (Applied
Biosystems Life Technologies, Foster City, CA, USA) with 5 ng of
cDNA, 1 µM of each primer pair and SYBR Green PCR master mix
(Roche). RT-qPCR was performed with the following primers: ET-1,
forward 5′-AGAGTGTGTCTACTTCTGCCA-3′ and reverse CTGCCGTCTAGAA-3′;
GAPDH, forward 5′-GGACCTGACCTGCCGTCTAGAA-3′ and reverse
5′-GGTGTCGCTGTTGAAGTCAGAG-3′. Relative mRNA levels were normalized
to GAPDH and quantified using the 2−ΔΔCq method
(25). Each experiment contained
three replicates and was repeated three times.
Assessment of eNOS activity
Cell lysates were prepared, as described for the
western blot analysis. The cell lysates were centrifuged at 14,000
× g for 10 min and the supernatant was discarded from the pelleted
cells. The activity of eNOS was detected by measuring the
conversion of [3H] l-arginine to [3H]
l-citrulline using an eNOS assay kit. (Cayman Chemical Co., Ann
Arbor, MI, USA), according to the manufacturer's protocol. The
results are expressed as a percentage of the control. Each
experiment contained three replicates and was repeated three
times.
MTS cell proliferation assays to assess
the toxicity of atorvastatin
The cells were seeded in 96-well plates (1,000
cells/well) and were treated with vehicle or with increasing doses
of atorvastatin (10−6, 10−5 and
10−4 M) for 24 and 48 h at 37°C with 5% CO2.
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxypheny
l)-2-(4-sulfophenyl)-2H-tetrazolium (Promega, Madison, WI, USA; 20
µl/well) was added for 2 h on the indicated days. Cell
proliferation was measured, according to the manufacturer's
protocol. Each experiment contained six replicates and was repeated
three times.
Statistical analysis
Data are presented as the mean ± standard deviation.
Differences between groups were analyzed using a two-tailed
unpaired t-test (GraphPad InStat; GraphPad Software, Inc.,
La Jolla, CA, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
High concentrations of glucose and
insulin reduce the protein expression levels of
phosphor(tyr)-IR and phosphor(tyr)-IRS-1,
production of NO, protein expression of
phosphor(ser1177)-eNOS and activity of eNOS, and
increase the mRNA level of ET-1 in HUVECs
To establish the effects of high glucose and high
insulin concentrations on the function of the HUVECs, the cells
were cultured in DMEM containing 5, 10 or 30 mM glucose, either
alone or in combination with 10−5 M insulin, for 24 h.
No differences in cell proliferation or cell morphology were
observed between the cells cultured in different concentrations of
glucose with or without 10−5 M insulin (data not shown).
The cells were then treated with insulin at an effective stimulus
concentration of 10−5 M for 6 h. As shown in Fig. 1A and B, 30 mM glucose significantly
decreased the protein levels of phosphor(tyr)-IR and
phosphor(tyr)-IRS-1 by 60 and 40%, respectively,
compared with the level in the cells cultured with 5 mM glucose. Of
note, when the cells were treated with the high dose (30 mM) of
glucose and 10−5 M insulin, there were further decreases
in phosphor(tyr)-IR and phosphor(tyr)-IRS-1
(P<0.05), compared with the cells treated with 30 mM glucose
alone. Specifically, the expression of phosphor(tyr)-IR
was reduced by 60% and the expression of
phosphor(tyr)-IRS-1 was reduced by 40%, compared with
the level in the cells cultured with 5 mM glucose. The addition of
10−5 M insulin alone did not alter the level of
phosphor(tyr)-IR, but decreased the level of
phosphor(tyr)-IRS-1 in the cells cultured with 5 mM
glucose.
 | Figure 1High concentrations of glucose and
insulin reduce the protein expression levels of
phosphor(tyr)-IR and phosphor(tyr)-IRS-1,
production of NO, protein expression of phosphor
(ser1177) -eNOS and activity of eNOS, and increase the
mRNA expression of ET-1 in HUVECs. The HUVECs were treated with
indicated concentrations of glucose in the absence (open bars) or
presence (filled bars) of insulin (10−5 M) for 24 h.
Western blot analysis of (A) phosphor (tyr) IR and (B)
phosphor (tyr) IRS-1. (C) Nitrate reduction assay of NO
concentrations and reverse transcription-quantitative polymerase
chain reaction analysis of ET-1 mRNA. ***P<0.001 vs.
media containing 5 mM glucose only. The values for ET-1 mRNA in 5
mM glucose without insulin are defined as 1 (##P<0.01
and ###P<0.001 vs. 1). (D) Western blot analysis of
the protein expression levels of phosphor (ser1177) eNOS
and total eNOS. (E) Assessment of eNOS activity. (A, B, D and E)
**P<0.01 vs. media containing 5 mM glucose only;
#P<0.05, ##P<0.01 vs. media containing
30 mM glucose only. The data are presented as the mean ± standard
deviation of three independent experiments. HUVECs, human umbilical
vein endothelial cells; phosphor/P, phosphorylated; t, total; IR,
insulin receptor; IRS-1, insulin receptor substrate-1; NO, nitric
oxide; eNOS, endothelial NO synthase. |
To confirm that the endothelial dysfunction was
induced by high levels of glucose and insulin, the total NO
concentration and mRNA level of ET-1 were measured. The protein
expression of eNOS was also evaluated. NO production was reduced in
the 30 mM glucose group (53.69±3.45 µM) compared with the 5
mM glucose group (116.17±4.93 µM), and 30 mM glucose
combined with 10−5 M insulin caused a further reduction
in NO production (36.26±5.24 µM; Fig. 1C). By contrast, the combination
treatment of 30 mM glucose and 10−5 M insulin induced an
increase of ~13 fold in the mRNA level of ET-1, compared with the
cells treated with 5 mM glucose.
The protein expression of eNOS was further assessed
in the present study. As shown in Fig.
1D, glucose and insulin treatment did not alter the expression
level of total eNOS, however, higher doses of glucose decreased the
protein expression of phosphor(ser1177)-eNOS, which was
further augmented by the presence of insulin. The phosphorylation
level of the cell cultured in 30 mM glucose and 10−5 M
insulin decreased to 22% of that of the cells cultured in 5 mM
glucose. It was also found that the activity of eNOS altered in
parallel with the protein expression of
phosphor(ser1177)-eNOS. The combination of 30 mM glucose
and 10−5 M insulin caused a significant reduction, to
25% of tn the cells cultured in 5 mM glucose (Fig. 1E).
Atorvastatin increases the protein
expression of phosphor(tyr)-IR and
phosphor(tyr)-IRS-1, production of NO, protein
expression of phosphor (ser1177)-eNOS and activity of
eNOS, and downregulates the mRNA level of ET-1 in a dose-dependent
manner
Prior to examining the effect of atorvastatin on the
production of NO, the present study recorded the effect of
atorvastatin on cell proliferation to assess the toxicity of the
atorvastatin. As shown in Fig. 2A,
no differences in cell proliferation were observed between the Nor
vec, IR vec and IR vec cells incubated with increasing doses of
atorvastatin for 48 h (P>0.05).
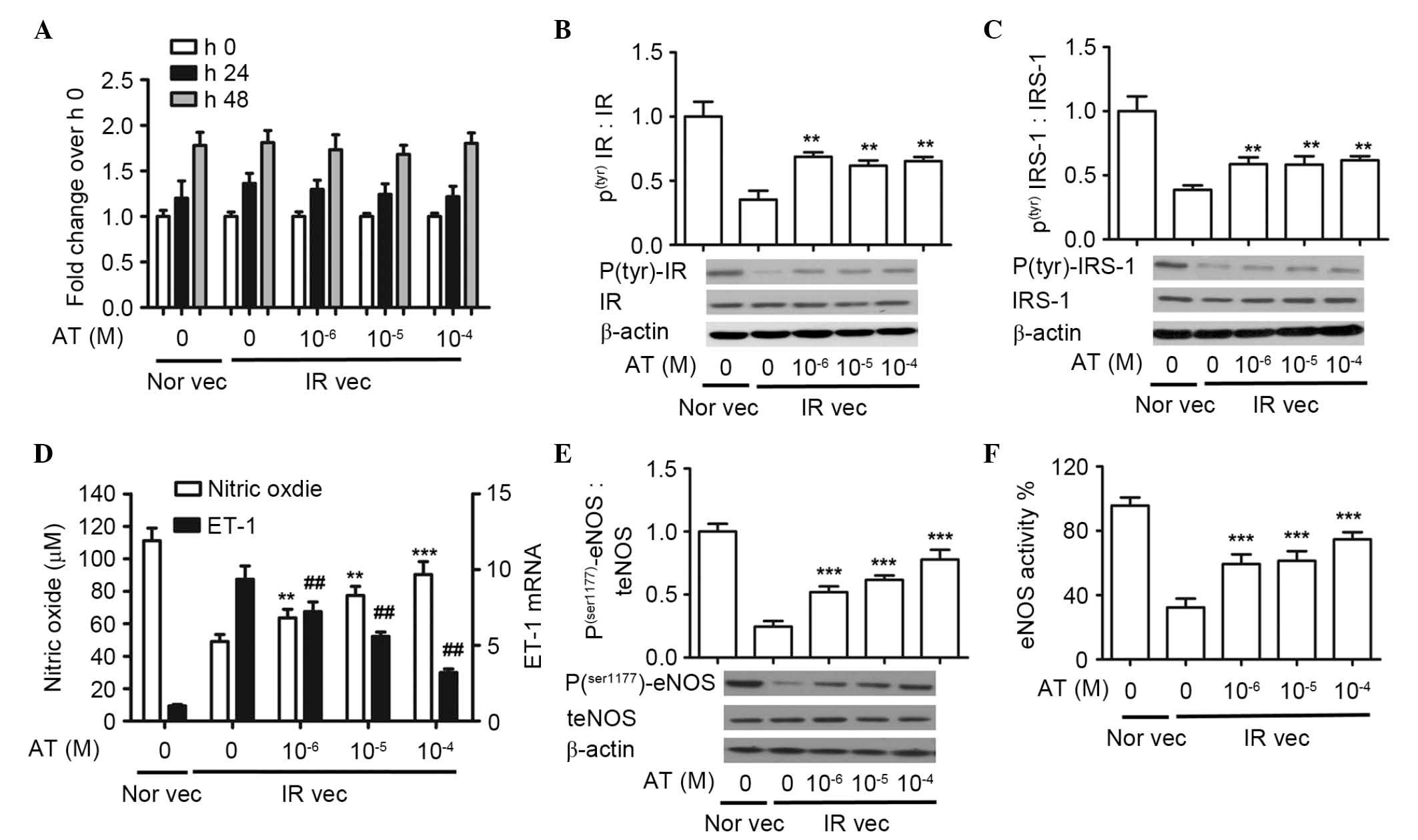 | Figure 2Atorvastatin increases the protein
expression of phosphor(tyr)-IR and
phosphor(tyr)-IRS-1, production of NO, protein
expression of phosphor (ser1177)-eNOS and activity of
eNOS, and downregulates the mRNA level of ET-1 in a dose-dependent
manner. The HUVECs were pretreated with 30 mM glucose and
10−5 M insulin, and then with atorvastatin (0,
10−6, 10−5 and 10−4 M). (A) Cell
proliferation rates were analyzed using an MTS assay. Western blot
analysis of (B) phosphor (tyr)IR and (C) phosphor
(tyr)IRS-1. (D) Nitrate reduction assay of NO
concentrations and reverse transcription-quantitative polymerase
chain reaction analysis of ET-1 mRNA. The values for ET-1 mRNA in
the IR vec cells without atorvastatin are defined as 1.
##P<0.01 vs. 1. (E) Western blot analysis of phosphor
(ser1177) eNOS and (F) assessment of eNOS activity.
(A-F) **P<0.01 and ***P<0.001 vs.
untreated IR vec. The data are presented as the mean ± standard
deviation of three independent experiments. Nor vec, cells cultured
in normal media; IR vec, cells cultured in media containing 30 mM
glucose and 10−5 M insulin; AT, atorvastatin; HUVECs,
human umbilical vein endothelial cells; phosphor/P, phosphorylated;
t, total; IR, insulin receptor; IRS-1, insulin receptor
substrate-1; NO, nitric oxide; eNOS, endothelial NO synthase. |
There was an apparent decrease in the
insulin-stimulated expression of phosphor(tyr)-IR in the
IR vec cells, which increased following treatment with
atorvastatin. There were significant differences in the expression
of phosphor(tyr)-IR between the Nor vec (1.00±0.07),
untreated IR vec (0.38±0.04) and IR vec cells treated with
increased doses of atorvastatin (10−6 M, 0.68±0.01;
10−5 M, 0.62±0.03; 10−4 M, 0.63±0.04). The
expression trend of phosphor(tyr)-IRS-1 was similar to
phosphor(tyr)-IR. There was a significant increase when
the IR vec cells were treated with increased doses of atorvastatin
(10−6 M, 0.57±0.05; 10−5 M, 0.55±0.05;
10−4 M, 0.60±0.03), compared with the untreated IR vec
cells (0.36±0.04). Increasing doses of atorvastatin did not alter
the protein expression levels of either phosphor
(tyr)-IR or phosphor (tyr)-IRS-1
(P>0.05).
The effect of atorvastatin on NO production was also
evaluated. As shown in Fig. 2D,
atorvastatin significantly increased NO production, compared with
the untreated IR vec group, in a dose dependent manner (untreated
IR vec, 49.10±4.23 µM; 10−6 M, 63.69±5.19
µM; 10−5 M, 77.43±5.56 µM; 10−4
M, 90.37±7.92 µM). By contrast, atorvastatin reduced the
mRNA expression of ET-1, compared with the untreated IR vec group.
Among the four treatment concentrations, the maximal inhibition
rate occurred at the dose of 10−4 M (76%), however,
inhibition was readily detectable at 10−6 M (24%).
Atorvastatin significantly increased the protein expression of
phosphor (ser1177)-eNOS in a dose-dependent manner,
compared with the untreated IR vec group: Untreated IR vec,
0.24±0.02; 10−6 M, 0.52±0.04; 10−5 M,
0.62±0.03; 10−4 M, 0.72±0.06). Atorvastatin
significantly increased the activity of eNOS, compared with the
untreated IR vec group: Untreated IR vec, 42.33±4.49%;
10−6 M, 61.67±5.43%; 10−5 M, 66.14±4.92%;
10−4 M, 74.67±3.68% (Fig.
2D and E).
Atorvastatin increases the production of
NO, protein expression of phosphor (ser1177)-eNOS and
activity of eNOS, and downregulates the mRNA expression of ET-1 in
a time-dependent manner
To further examine the effect of atorvastatin, the
levels of NO produced in the Nor vec and IR vec cells were measured
every 3 h. As shown in Fig. 3A, in
the IR vec cells, atorvastatin induced the production of NO, the
level of which was highest at 9 h and was maintained during the
duration of the asssessment. By contrast no significant alterations
were observed in the Nor vec cells. Atorvastatin caused a reduction
in the mRNA levels of ET-1 in the Nor vec and IR vec cells. The
effect of atorvastatin on the activity of eNOS was also measured.
As shown in Fig. 3C, atorvastatin
did not alter the expression level of total eNOS in either the Nor
vec and IR vec cells during the assessment, however, atorvastatin
increased the protein expression of phosphor
(ser1177)-eNOS in a time dependent manner. In addition,
eNOS activity in the IR vec cells increased following atorvastatin
treatment, but remained at the same level in the Nor vec cells in
24 h.
 | Figure 3Atorvastatin increases the production
of NO, the protein expression of phosphor (ser1177)
-eNOS and the activity of eNOS, and downregulates the mRNA
expression of ET-1 in a time-dependent manner. HUVECs were cultured
in normal media (Nor vec) and pretreated with 30 mM glucose and
10−5 M insulin for 24 h (IR vec) with or without
10−4 M atorvastatin (A) Nitrate reduction assay of NO
concentrations and (B) reverse transcription-quantitative
polymerase chain reaction analysis of ET-1 mRNA were performed
every 3 h. (C) Western blot analysis of protein levels of phosphor
(ser1177)-eNOS and total eNOS; (D) eNOS activity was
measured at the indicated time points. The data are presented as
the mean ± standard deviation of three independent experiments.
*P<0.05 and **P<0.01 vs. IR vec at 0 h.
AT, atorvastatin; HUVECs, human umbilical vein endothelial cells;
phosphor/P, phosphorylated; t, total; IR, insulin receptor; IRS-1,
insulin receptor substrate-1; NO, nitric oxide; eNOS, endothelial
NO synthase. |
LY29004, a PI3K inhibitor, decreases the
effect of atorvastatin on NO production, protein expression of
phosphor (ser1177)-eNOS and activity of eNOS, but not
the mRNA level of ET-1
To determine whether atorvastatin stimulates eNOS
activation in a PI3K/Akt/eNOS-dependent manner, the cells were
treated with the PI3K inhibitor, LY294002. As shown in Fig. 4A, atorvastatin induced an increase
in the production of NO in the Nor vec and IR vec cells. This
effect was reduced by LY294002 (P<0.05). Similarly, the effects
of atorvastatin on the protein expression of phosphor
(ser1177)-eNOS and activity of eNOS were reduced by LY294002 in the
Nor vec and IR vec cells (P<0.05; Fig. 4B and C). However, the redutcion
observed in the mRNA expression of ET-1 was not affected by
LY294002 (P>0.05).
 | Figure 4PI3K inhibitor,LY29004, decreases the
effect of atorvastatin on NO production, the protein expression of
phosphor (ser1177)-eNOS and activity of eNOS, but does
not affect the mRNA level of ET-1. HUVECs cultured in normal media
(Nor vec) and pretreated with 30 mM glucose and 10−5 M
insulin media (IR vec) were treated with PBS (control),
10−4 M atorvastatin, 25 µM LY29004, or a
combination of 10−4 M atorvastatin and 25 µM
LY29004, respectively. (A) Nitrate reduction assay of NO
concentrations; (B) western blot analysis of the protein expression
levels of phosphor (ser1177) eNOS and total eNOS; (C)
assessment of eNOS activity, (D) reverse transcription-quantitative
polymerase chain reaction analysis of ET-1 mRNA. The data are
presented as the mean ± standard deviations, *P<0.05,
**P<0.01 and ***P<0.001 vs. IR vec
treated with PBS; #P<0.05 and ##P<0.01
vs. Nor vec treated with PBS. PI3K, phosphatidylinositol 3-kinase;
AT, atorvastatin HUVECs, human umbilical vein endothelial cells;
phosphor/P, phosphorylated; t, total; IR, insulin receptor; IRS-1,
insulin receptor substrate-1; NO, nitric oxide; eNOS, endothelial
NO synthase; CON, control. |
Discussion
Impairment of vascular endothelial cell structure
and function is a common pathological basis of cardiovascular
disease. The predominant factors, which lead to vascular
endothelial dysfunction include dyslipidemia, impairment of
endothelium-dependent vasodilation and inflammation (1). Insulin resistance, the early stage of
type 2 diabetes, is frequently associated with endothelial
dysfunction as an early predictor of atherosclerosis and risk of
cardiovascular disease (13,26).
However, the mechanism of interaction between insulin resistance
and endothelial dysfunction remains to be elucidated. Atorvastatin
is a vessel protective drug, which has been confirmed to protect
endothelial function by increasing NO production, and reducing
circulating levels of interleukin (IL)-6 and tumor necrosis-factor
(TNF)-α independent of changes in plasma cholesterol (27,28).
In the present study, an insulin resistant-endothelial dysfunction
model was established by treating the HUVECs with high
concentrations of glucose and insulin for 24 h. Compared with
normal culture media, high concentrations of glucose and insulin
interfered with insulin signaling by decreasing insulin-stimulated
phosphor (tyr)-IR and downstream IRS-1. This result is
consistent with previous reports that hyperglycemia can induce
insulin resistance by inhibiting phosphor (tyr)-IRS-1,
which in turn negatively regulates its function (29,30).
In individuals with insulin-resistance, the
impairment of insulin signal transduction via the PI3K pathway can
downregulate the level of phosphor (ser1177)-eNOS and
decrease NO production (31). In
the present study, impairment of endothelial insulin signaling was
accompanied by a reduction in tyrosine phosphorylation of the
insulin receptor substrate. Previous reports have indicated that
the expression of TNF-α is significantly increased in insulin
resistance by decreasing the tyrosine kinase activity of the
insulin receptor (32,33). In addition, TNF-α induces the
activation of NAD(P)H oxidase, leading to endothelial dysfunction
with increased ET-1 availability (34,35).
Thus, the present study hypothesized that superphysiological
concentrations of insulin and glucose increase the expression of
ET-1 through the downstream effects of the upregulation of TNF-α on
NAD(P)H oxidase and superoxide anion production. The effects of
atorvastatin on insulin sensitivity have been reported in previous
years. Wong et al reported that atorvastatin induced insulin
sensitization in Zucker lean and fatty rats (36). Atorvastatin also reverses the
reduction of phosphor (tyr)-IR and phosphor
(tyr)-IRS-1 in animal models of insulin resistance or
impairment of insulin signaling (37,38).
However, the function of atorvastatin on insulin sensitivity in
insulin resistant-endothelial dysfunction cell models has not been
reported. The data obtained in the present study are the first, to
the best of our knowledge, to show that atorvastatin reversed the
inhibition of phosphor (tyr)-IR and phosphor
(tyr)-IRS-1 induced by high glucose in combination with
high insulin. In addition, atorvastatin increased the production of
NO and dowregulated the expression of ET-1 in a dose and time
dependent manner.
In the present study, the PI3K inhibitor, Ly294002,
was used. The production of NO, protein expression of phosphor
(ser1177)-eNOS and activity of eNOS were significantly
decreased when the cells were treated with LY294002, which
indicated that PI3K was a specific upstream effector to
phosphorylate the eNOS on ser1177. In conclusion, the present study
provided the first evidence, to the best of our knowledge, that a
high concentration of glucose in combination with a high
concentration of insulin stimulated endothelial insulin resistance
in vascular endothelial cells. In addition, atorvastatin
ameliorated these effects, primarily via the PI3K/Akt/eNOS pathway.
These findings provide further evidence that atorvastatin is useful
for patients with insulin resistance.
Acknowledgments
This study was supported by grants from the
Scientific Research Project of the Department of Finance, Jilin
Province, China (grant no. 3D512W143428). The authors would like to
thank Dr. William Orr, Department of Pathology, University of
Manitoba, Canada for his assistance in manuscript preparation.
References
|
1
|
Versari D, Daghini E, Virdis A, Ghiadoni L
and Taddei S: Endothelial dysfunction as a target for prevention of
cardiovascular disease. Diabetes Care. 32(Suppl 2): S314–S321.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cersosimo E and DeFronzo RA: Insulin
resistance and endothelial dysfunction: The road map to
cardiovascular diseases. Diabetes Metab Res Rev. 22:423–436. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Davignon J and Ganz P: Role of endothelial
dysfunction in atherosclerosis. Circulation. 109(23 Suppl 1):
IIII27–III32. 2004. View Article : Google Scholar
|
|
4
|
Reaven GM: Banting lecture 1988. Role of
insulin resistance in human disease. Diabetes. 37:1595–1607. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wheatcroft SB, Williams IL, Shah AM and
Kearney MT: Pathophysiological implications of insulin resistance
on vascular endothelial function. Diabetic Med. 20:255–268. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heitzer T, Schlinzig T, Krohn K, Meinertz
T and Münzel T: Endothelial dysfunction, oxidative stress and risk
of cardiovascular events in patients with coronary artery disease.
Circulation. 104:2673–2678. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ginsberg HN: Insulin resistance and
cardiovascular disease. J Clin Invest. 106:453–458. 2000.
View Article : Google Scholar
|
|
8
|
Kubota T, Kubota N, Kumagai H, Yamaguchi
S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, et
al: Impaired insulin signaling in endothelial cells reduces
insulin-induced glucose uptake by skeletal muscle. Cell Metab.
13:294–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rask-Madsen C, Li Q, Freund B, Feather D,
Abramov R, Wu IH, Chen K, Yamamoto-Hiraoka J, Goldenbogen J,
Sotiropoulos KB, et al: Loss of insulin signaling in vascular
endothelial cells accelerates atherosclerosis in apolipoprotein E
null mice. Cell Metab. 11:379–389. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim F, Pham M, Maloney E, Rizzo NO, Morton
GJ, Wisse BE, Kirk EA, Chait A and Schwartz MW: Vascular
inflammation, insulin resistance and reduced nitric oxide
production precede the onset of peripheral insulin resistance.
Arterioscler Thromb Vasc Biol. 28:1982–1988. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Okamoto H, Obici S, Accili D and Rossetti
L: Restoration of liver insulin signaling in Insr knockout mice
fails to normalize hepatic insulin action. J Clin Invest.
115:1314–1322. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garcia Soriano F, Virág L, Jagtap P, Szabó
E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL,
et al: Diabetic endothelial dysfunction: The role of
poly(ADP-ribose) polymerase activation. Nat Med. 7:108–113. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rask-Madsen C and King GL: Mechanisms of
disease: Endothelial dysfunction in insulin resistance and
diabetes. Nat Clin Pract Endocrinol Metab. 3:46–56. 2007.
View Article : Google Scholar
|
|
14
|
Arcaro G, Cretti A, Balzano S, Lechi A,
Muggeo M, Bonora E and Bonadonna RC: Insulin causes endothelial
dysfunction in humans sites and mechanisms. Circulation.
105:576–582. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burén J, Liu HX, Lauritz J and Eriksson
JW: High glucose and insulin in combination cause insulin receptor
substrate-1 and-2 depletion and protein kinase B desensitisation in
primary cultured rat adipocytes: Possible implications for insulin
resistance in type 2 diabetes. Eur J Endocrinol. 148:157–167. 2003.
View Article : Google Scholar
|
|
16
|
Gelosa P, Cimino M, Pignieri A, Tremoli E,
Guerrini U and Sironi L: The role of HMG-CoA reductase inhibition
in endothelial dysfunction and inflammation. Vasc Health Risk
Manag. 3:567–577. 2007.PubMed/NCBI
|
|
17
|
Durazzo AE, Machado FS, Ikeoka DT, De
Bernoche C, Monachini MC, Puech-Leão P and Caramelli B: Reduction
in cardiovascular events after vascular surgery with atorvastatin:
A randomized trial. J Vasc Surg. 39:967–975; discussion 975–976.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chello M, Goffredo C, Patti G, Candura D,
Melfi R, Mastrobuoni S, Di Sciascio G and Covino E: Effects of
atorvastatin on arterial endothelial function in coronary bypass
surgery. Eur J Cardiothorac Surg. 28:805–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dilaveris P, Giannopoulos G, Riga M,
Synetos A and Stefanadis C: Beneficial effects of statins on
endothelial dysfunction and vascular stiffness. Curr Vasc
Pharmacol. 5:227–237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mahmoud MF, El-Nagar M and El-Bassossy HM:
Anti-inflammatory effect of atorvastatin on vascular reactivity and
insulin resistance in fructose fed rats. Arch Pharm Res.
35:155–162. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Calisto KL, Carvalho B, Ropelle ER,
Mittestainer FC, Camacho AC, Guadagnini D, Carvalheira JB and Saad
MJ: Atorvastatin improves survival in septic rats: Effect on tissue
inflammatory pathway and on insulin signaling. PLoS One.
5:e142322010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Shi Y, Wei Y, Ma X, Li Y and Li R:
Altered expression profiles of microRNAs upon arsenic exposure of
human umbilical vein endothelial cells. Environ Toxicol Pharmacol.
34:381–387. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schmidt RJ and Baylis C: Total nitric
oxide production is low in patients with chronic renal disease.
Kidney Int. 58:1261–1266. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Lebovitz HE: Insulin resistance-a common
link between type 2 diabetes and cardiovascular disease. Diabetes
Obes Metab. 8:237–249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tousoulis D, Antoniades C, Katsi V,
Bosinakou E, Kotsopoulou M, Tsioufis C and Stefanadis C: The impact
of early administration of low-dose atorvastatin treatment on
inflammatory process, in patients with unstable angina and low
cholesterol level. Int J Cardiol. 109:48–52. 2006. View Article : Google Scholar
|
|
28
|
Tousoulis D, Charakida M, Stefanadi E,
Siasos G, Latsios G and Stefanadis C: Statins in heart failure.
Beyond the lipid lowering effect. Int J Cardiol. 115:144–150. 2007.
View Article : Google Scholar
|
|
29
|
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi
NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH and Hotamisligil
GS: Endoplasmic reticulum stress links obesity, insulin action, and
type 2 diabetes. Science. 306:457–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hribal ML, Perego L, Lovari S, Andreozzi
F, Menghini R, Perego C, Finzi G, Usellini L, Placidi C, Capella C,
et al: Chronic hyperglycemia impairs insulin secretion by affecting
insulin receptor expression, splicing and signaling in RIN beta
cell line and human islets of Langerhans. FASEB J. 17:1340–1342.
2003.PubMed/NCBI
|
|
31
|
Muniyappa R and Quon MJ: Insulin action
and insulin resistance in vascular endothelium. Curr Opin Clin Nutr
Metab Care. 10:523–530. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gonzalez-Gay MA, De Matias JM,
Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J
and Llorca J: Anti-tumor necrosis factor-alpha blockade improves
insulin resistance in patients with rheumatoid arthritis. Clin Exp
Rheumatol. 24:83–86. 2006.PubMed/NCBI
|
|
33
|
De Taeye BM, Novitskaya T, McGuinness OP,
Gleaves L, Medda M, Covington JW and Vaughan DE: Macrophage
TNF-alpha contributes to insulin resistance and hepatic steatosis
in diet-induced obesity. Am J Physiol Endocrinol Metab.
293:E713–E725. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
López-Sepúlveda R, Gómez-Guzmán M,
Zarzuelo MJ, Romero M, Sánchez M, Quintela AM, Galindo P, O'Valle
F, Tamargo J, Pérez-Vizcaíno F, et al: Red wine polyphenols prevent
endothelial dysfunction induced by endothelin-1 in rat aorta: Role
of NADPH oxidase. Clin Sci (Lond). 120:321–333. 2011. View Article : Google Scholar
|
|
35
|
Zhang H, Zhang J, Ungvari Z and Zhang C:
Resveratrol Improves endothelial function: Role of TNF{alpha} and
vascular oxidative stress. Arterioscler Thromb Vasc Biol.
29:1164–1171. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wong V, Stavar L, Szeto L, Uffelman K,
Wang CH, Fantus IG and Lewis GF: Atorvastatin induces insulin
sensitization in Zucker lean and fatty rats. Atherosclerosis.
184:348–355. 2006. View Article : Google Scholar
|
|
37
|
Lalli CA, Pauli JR, Prada PO, Cintra DE,
Ropelle ER, Velloso LA and Saad MJ: Statin modulates insulin
signaling and insulin resistance in liver and muscle of rats fed a
high-fat diet. Metabolism. 57:57–65. 2008. View Article : Google Scholar
|
|
38
|
Calisto KL, Carvalho Bde M, Ropelle ER,
Mittestainer FC, Camacho AC, Guadagnini D, Carvalheira JB and Saad
MJ: Atorvastatin improves survival in septic rats: Effect on tissue
inflammatory pathway and on insulin signaling. PloS One.
5:e142322010. View Article : Google Scholar : PubMed/NCBI
|














