Introduction
Dilated cardiomyopathy (DCM) is a heart muscle
disease characterized by ventricular chamber enlargement and
contractile dysfunction with normal left ventricular wall thickness
(1). It is the most common form of
primary cardiomyopathy, affecting approximately 1 in 2,500
individuals worldwide (1). DCM may
give rise to congestive heart failure, ventricular and
supraventricular arrhythmias, cardiac conduction blocks,
thromboembolism and sudden cardiac death. DCM is the third most
common cause of heart failure and the most frequent reason for
requiring heart transplantation (1,2).
Accumulating evidence demonstrates that genetic risk factors serve
a crucial role in the pathogenesis of familial DCM, which can be
inherited in an autosomal dominant, autosomal recessive, X-linked,
or mitochondrial pattern, with the pattern of autosomal dominant
inheritance present in greater than 90% of patients (3,4). At
present, >50 genes have been associated with familial or
sporadic DCM (4–9). The majority of these DCM-associated
genes encodes for sarcomeric proteins in addition to cytoskeletal
proteins, including contractile elements, sarcolemma elements,
Z-disc elements and Z-I region components, which are responsible
for the generation and transmission of contractile force (4,7).
However, these known DCM-causing genes only account for
approximately a third of patients with DCM and furthermore, the
majority of these have a low mutation frequency, occurring in
<1% of patients with DCM (10).
Thus, the identification of novel genetic defects underpinning DCM
is required in order to fully elucidate the molecular mechanisms of
DCM.
Previously, numerous key cardiac transcription
factors, including homeobox protein NK2 homeobox 5 (NKX2-5), zinc
finger proteins GATA binding protein 4 (GATA4), GATA5 and GATA6,
and T-box factors T-Box 5 (TBX5) and TBX20, have been demonstrated
to physically interact with each other to mediate embryonic
cardiogenesis and adult cardiac remodeling (11–18).
In addition, mutations in these core cardiac transcription factors
have been causally associated with various congenital heart
diseases (CHD) and cardiac arrhythmias (19–40).
Of note, mutations in NKX2-5, GATA4, GATA5, GATA6 and TBX5 have
additionally been associated with DCM in humans (41–49).
Given that the expression profile and functional characteristics of
TBX20 overlap at least partially with those of
NKX2-5, GATA4, GATA5, GATA6 and
TBX5 (13–15,18,50),
it is suggested that mutations in TBX20 may contribute to
DCM in a subset of patients.
Materials and methods
Study subjects
In the current case-control study, 115 unrelated
patients (65 males and 50 females, mean age of 52 years) with
idiopathic DCM were enrolled from a Han Chinese population, with
400 ethnically-matched (228 males and 172 females, mean age of 52
years), unrelated healthy individuals recruited as controls. The 14
available family members of the index patient carrying an
identified TBX20 mutation were also included. The
participants of the present study were recruited from Yangpu
Hospital, Renji Hospital, Shanghai Jiao Tong University and
Shanghai Chest Hospital (Shanghai, China), during between January
2013 and December 2014. All study participants were clinically
evaluated by medical history, physical examination,
echocardiography, electrocardiogram and exercise performance
testing. Cardiac catheterization, coronary angiography, myocardial
biopsy or cardiac magnetic resonance imaging were performed only if
there was a strong indication for the corresponding procedure.
Diagnosis of idiopathic DCM was made as described previously: Left
ventricular end-diastolic diameter >27 mm/m2 and an
ejection fraction <40% or fractional shortening <25% in the
absence of underlying conditions including ischemic heart disease,
hypertension, CHD, valvular heart disease, myocarditis and
metabolic disorders (42–49). The current study was conducted in
line with the principles of the Declaration of Helsinki. The study
protocol was approved by the ethics committee of Yangpu Hospital
(Tongji University School of Medicine, Shanghai, China) and written
informed consent was obtained by all participants prior to
investigation.
Genetic scan of TBX20
Peripheral venous blood samples were obtained by
using syringe from antecubital veins of all study subjects. Genomic
DNA was isolated from blood cells with the Wizard Genomic DNA
Purification kit (Promega Corporation, Madison, WI, USA). The
coding exons and flanking introns of the TBX20 gene were
sequenced in 115 unrelated patients with sporadic DCM, in addition
to in 200 control individuals. The primer pairs used to genotype
TBX20 by polymerase chain reaction (PCR)-sequencing were
designed as previously described (32). PCR was performed using HotStar Taq
DNA Polymerase (Qiagen GmbH, Hilden, Germany) on a Veriti Thermal
Cycler (Applied Biosystems; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with standard conditions and concentrations of
reagents (27). Both strands of
each PCR product were sequenced with a BigDye®
Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) on an ABI PRISM 3130 XL DNA Analyzer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). DNA sequences
were analyzed with DNA Sequencing Analysis software, version 5.1
(Applied Biosystems; Thermo Fisher Scientific, Inc.). A variant was
confirmed by re-sequencing of an independent PCR-generated amplicon
from the same subject. For each identified sequence variant, the
single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) database and human
gene mutation database (HGMD; http://www.hgmd.org) were searched for its
novelty.
Prediction of the disease-causing
potential of a TBX20 sequence variation
The causative potential of a TBX20 sequence
variation was predicted using MutationTaster (http://www.mutationtaster.org), which produced the
probability for the variation to be either a pathogenic mutation or
a benign polymorphism. Notably, the given P-value was the
probability of the correct prediction rather than the probability
of error as used in t-test statistics. Thus, a value close to 1
indicates a high accuracy of prediction.
Expression plasmids and site-directed
mutagenesis
The recombinant expression plasmid TBX20-pcDNA3.1
was constructed as described previously (32). The expression plasmids GATA4-pSSRa
and NKX2-5-pEFSA, and the reporter plasmid ANF-luciferase
(ANF-luc), which contains the 2600-bp 5′-flanking region of the
ANF gene and expresses Firefly luciferase, were provided by
Dr Ichiro Shiojima from Chiba University School of Medicine (Chiba,
Japan). The identified mutation was introduced into the wild-type
TBX20-pcDNA3.1 construct by site-directed mutagenesis using the
QuikChange II XL Site-Directed Mutagenesis kit (Stratagene; Agilent
Technologies, Inc., Santa Clara, CA, USA) and a complementary pair
of primers (F: 5′-TCGGGGGTGGATCCTTAGGCCAAGTACATAG-3′; R:
5′-CTATGTACTTGGCCTAAGGATCCACCCCCGA-3′). The mutant was sequenced to
confirm the desired mutation and to exclude any other sequence
variations.
Cell culture and reporter gene
assays
COS-7 cells were grown in 6-well plates in
Dulbecco's modified Eagle's medium supplemented with 10% fetal calf
serum and maintained in an atmosphere of 10% CO2 at
37°C. Transfections of COS-7 cells were conducted at approximately
90% confluency with Lipofectamine™ 2000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). In cell transfection experiments, 0.8
µg wild-type TBX20-pcDNA3.1, 0.8 µg E143X-mutant
TBX20-pcDNA3.1, 0.4 µg wild-type TBX20-pcDNA3.1 or 0.4
µg wild-type TBX20-pcDNA3.1 together with 0.4 µg
E143X-mutant TBX20-pcDNA3.1 was used, in combination with 1.0
µg ANF-luc and 0.04 µg pGL4.75 (Promega Corporation).
In order to investigate the ability of the mutant TBX20 to activate
the ANF gene synergistically with NKX2-5 or GATA4, the same
quantity (0.6 µg) of expression plasmid DNA (empty pcDNA3.1,
wild-type TBX20-pcDNA3.1, NKX2-5-pEFSA, GATA4-pSSRa or E143X-mutant
TBX20-pcDNA3.1) was used alone or together, in the presence of 1.0
µg ANF-luc and 0.04 µg pGL4.75. The empty expression
vector plasmid pcDNA3.1 was used as a negative control. The
internal control reporter vector pGL4.75 expressing Renilla
luciferase was used to normalize transfection efficiency. Cells
were harvested and lysed 36 h subsequent to transfection.
Luciferase assays were performed using a Dual-Glo luciferase assay
kit (Promega Corporation) according to the manufacturer's protocol.
The results were expressed as fold activity of Firefly luciferase
relative to Renilla luciferase. A total of 14 separate transfection
experiments were conducted, and for each experiment, transfections
were performed in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviation.
Continuous variables were compared between two groups using
student's unpaired t-test. Comparison of the categorical variables
between two groups was performed using Pearson's χ2
test. Two-tailed P<0.05 was considered to indicate a
statistically significant difference.
Results
Baseline clinical characteristics of the
study subjects
A cohort of 115 unrelated patients with sporadic DCM
underwent clinical investigation in contrast to a total of 400
unrelated control individuals. None of them had other identifiable
risk factors for DCM. All the patients developed DCM when they were
reached adulthood. The average age of onset of DCM was 43±8 years,
with a median of 43 years. The control individuals had no evidence
of cardiac structural abnormality or functional impairment, and
their echocardiographic results were normal. There was no
significant difference in the age, gender and body-mass index
between the case and control groups (P>0.05). Compared with
those in the control group, in the patient group the arterial blood
pressure levels and left ventricular ejection fraction were
significantly reduced (P<0.05), whereas the heart rate, left
ventricular end-diastolic diameter and left ventricular
end-systolic diameter were significantly increased (P<0.05). The
baseline clinical characteristics of the study subjects are
summarized in Table I.
 | Table IBaseline clinical characteristics of
the patients with DCM and control individuals. |
Table I
Baseline clinical characteristics of
the patients with DCM and control individuals.
| Parameters | Patients
(n=115) | Controls
(n=400) | P-value |
|---|
| Age (years) | 52±14 | 52±12 | 1.0000 |
| Male | 65 (57) | 228 (57) | 0.9273 |
| BMI
(kg/m2) | 23±5 | 23±6 | 1.0000 |
| SBP (mmHg) | 116±17 | 128±15 | <0.0001 |
| DBP (mmHg) | 72±10 | 84±9 | <0.0001 |
| HR (bpm) | 82±15 | 74±11 | <0.0001 |
| LVEDD (mm) | 68±8 | 47±6 | <0.0001 |
| LVESD (mm) | 55±6 | 34±5 | <0.0001 |
| LVEF (%) | 37±11 | 62±8 | <0.0001 |
| NYHA function
class |
| I | 11 (10) | NA | NA |
| II | 29 (25) | NA | NA |
| III | 52 (45) | NA | NA |
| IV | 23 (20) | NA | NA |
| Medicine |
| ACEI | 71 (62) | NA | NA |
| ARB | 22 (19) | NA | NA |
| β-blockers | 67 (58) | NA | NA |
| Diuretics | 95 (83) | NA | NA |
| Digitalis | 86 (75) | NA | NA |
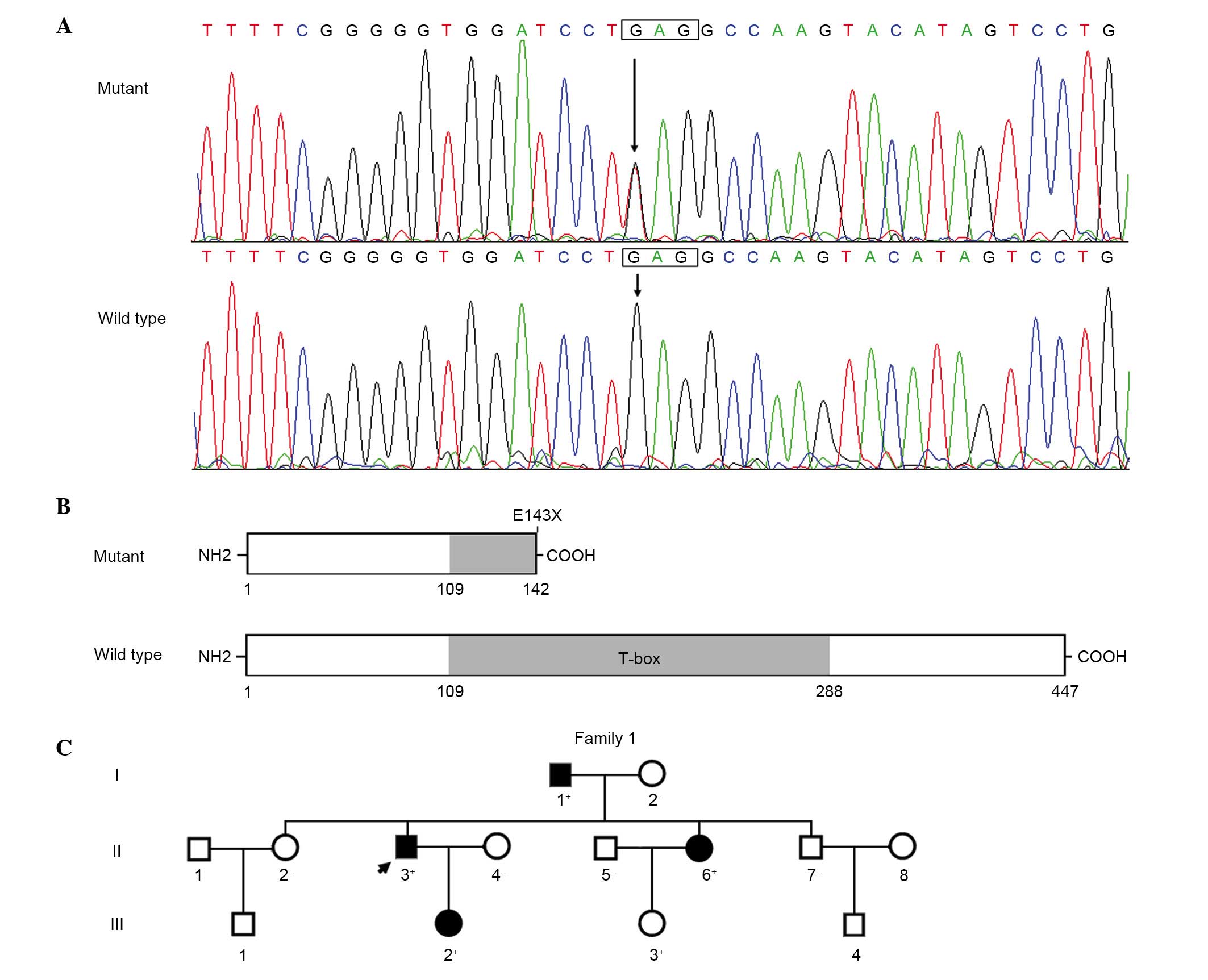
TBX20 mutation
By direct sequencing of TBX20, a heterozygous
mutation was identified in one of 115 unrelated patients with
sporadic DCM, with a mutational prevalence of nearly 0.87%.
Specifically, a transition of guanine (G) to thymine (T) in the
first nucleotide of codon 143 (c.427G>T), resulting in a
substitution of stop codon for glutamic acid codon at amino acid
position 143 (p.E143X), was identified in an index patient. The
sequence chromatograms demonstrating the heterozygous TBX20
mutation and its wild-type control sequence are presented in
Fig. 1A. A schematic diagram of
the TBX20 protein depicting the structural domains and location of
the mutation identified in the present study is presented in
Fig. 1B. The nonsense mutation was
neither observed in the 400 control individuals nor in the SNP and
HGM databases. Genetic screening of the proband's available family
members demonstrated that the mutation was present in all family
members affected with DCM, which was transmitted in an autosomal
dominant pattern in the family. However, all the DCM-affected
family members appeared healthy when they were juveniles.
Additionally, the proband's sister (II-6) additionally presented
with congenital atrial septal defect (ASD) and her juvenile
daughter (III-3) only presented with ASD. The pedigree structure of
the family is illustrated in Fig.
1C. The phenotypic characteristics and mutation status of the
affected family members are presented in Table II.
| Table IIPhenotypic characteristics and TBX20
mutation status of the affected family members. |
Table II
Phenotypic characteristics and TBX20
mutation status of the affected family members.
| Individual | Gender | Age
(years) | Cardiac
phenotype | LVEDD
(mm) | LVESD
(mm) | LVEF
(%) | LVFS
(%) | ECG findings |
TBX20mutation
(E143X) |
|---|
| I-1 | M | 68 | DCM | 56 | 65 | 26 | 14 | AVB, PVC | +/− |
| II-3 | M | 44 | DCM | 45 | 58 | 32 | 23 | | +/− |
| II-6 | F | 39 | DCM, ASD | 47 | 55 | 36 | 15 | AVB | +/− |
| III-2 | F | 22 | DCM | 43 | 54 | 48 | 21 | | +/− |
| III-3 | F | 14 | ASD | 37 | 25 | 65 | 32 | IRBBB | +/− |
Disease-causing potential of the TBX20
variation
The TBX20 sequence variation of c.427G>T
was predicted by MutationTaster to be a pathogenic mutation, with a
P-value of 1.
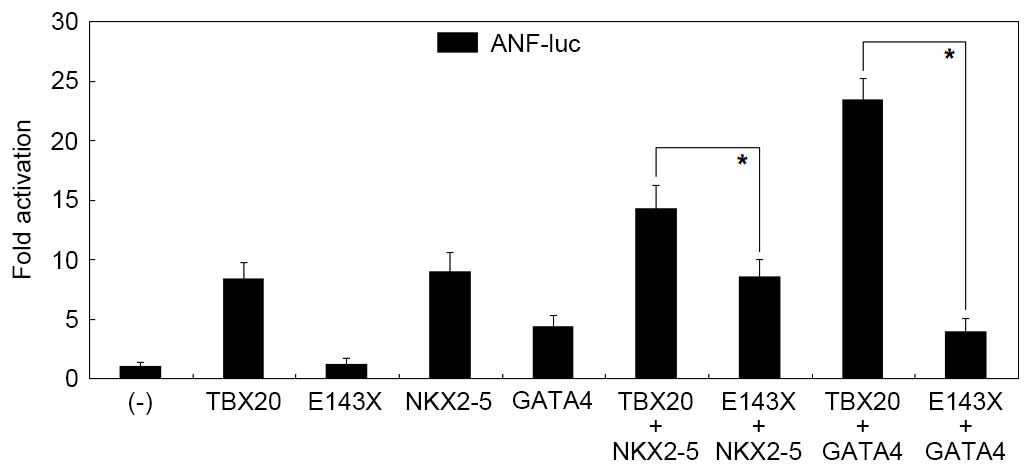
Diminished transcriptional activity of
the mutant TBX20 protein
As presented in Fig.
2, the same amount (0.8 µg) of wild-type TBX20 and
E143X-mutant TBX20 activated the ANF promoter by ~11-fold
and ~1-fold, respectively (wild type vs. mutant: t=11.7530,
P=0.0003). When 0.4 µg wild-type TBX20 was transfected alone
or together with 0.4 µg E143X-mutant TBX20, the induced
activation of the ANF promoter was ~6-fold (wild type vs.
wild type plus mutant: t=4.5758, P=0.0102). These results
demonstrate that the mutant TBX20 lacks transactivational activity
and has no dominant-negative effect on wild-type TBX20.
Disrupted synergistic transactivation
between mutant TBX20 and NKX2-5 or GATA4
As presented in Fig.
3, in the presence of 0.6 µg wild-type NKX2-5, the same
amount (0.6 µg) of wild-type and E143X-mutant TBX20
activated the ANF promoter by ~14-fold and ~9-fold,
respectively (wild type vs. mutant in the presence of NKX2-5:
t=4.1650, P=0.0141); while in the presence of 0.6 µg
wild-type GATA4, the same amount (0.6 µg) of wild-type and
E143X-mutant TBX20 activated the ANF promoter by ~23-fold
and ~4-fold, respectively (wild type vs. mutant in the presence of
GATA4: t=16.3428, P<0.0001). These data indicate that the
E143X-mutant TBX20 fails to transactivate the ANF promoter
in synergy with NKX2-5 or GATA4.
Discussion
In the current study, a novel heterozygous TBX20
mutation of p.E143X was identified in a family with DCM or ASD. The
nonsense mutation, which was predicted to be causative by
MutationTaster, was absent in the 800 reference chromosomes from an
ethnically-matched control population. Functional analyses
demonstrated that the mutant TBX20 failed to transcriptionally
activate the ANF promoter and exhibited no inhibitory effect
on its wild-type counterpart. Furthermore, the mutation disrupted
the synergistic activation between TBX20 and NKX2-5 or GATA4. These
observations suggest that haploinefficiency resulting from the
TBX20 mutation is an alternative mechanism of DCM or CHD in
a subset of patients.
In order to maintain the high fidelity of gene
expression, cells may utilize multiple decay approaches to
eliminate nonfunctional transcripts. At the mRNA level, there are
three avenues to protect cells from the possible accumulation of
abnormal mRNA and potentially toxic proteins, including non-stop
decay, which detects and degrades mRNAs lacking a stop codon, no-go
decay, which targets mRNAs with ribosomes arrested in translation
elongation, and nonsense-mediated mRNA decay (NMD), which promotes
the degradation of mRNAs undergoing premature translation
termination (51). The premature
termination codon (PTC) is derived from various types of mutations,
including nonsense mutations that convert a sense codon into an
in-frame PTC, insertion or deletion mutations that alter the
ribosomal reading frame, causing translating ribosomes to encounter
a PTC, and mutations that result in mRNA splicing defects that lead
to retention of an intron altering the reading frame, causing
translating ribosomes to encounter a PTC (52). In the current study, the identified
nonsense mutation in TBX20 is suggested to induce NMD,
resulting in haploinefficiency. However, due to inaccessibility to
the mutation carriers' tissue specimens, it was not possible to
confirm the absence of the truncated TBX20 protein.
Multiple TBX20 mutations have been previously
associated with enhanced vulnerability to DCM in humans. Kirk et
al (53) sequenced
TBX20 in 352 unrelated patients with CHD and identified two
mutations (p.I152 M and p.Q195X) in two index patients with ASD,
respectively. Genetic analysis of the two pedigrees indicated that
in the family harboring Q195X mutation, there were two living
mutation carriers, of whom a female had DCM and mitral valve
malformation, and a male had DCM, ASD, coarctation of the aorta and
pulmonary hypertension. Functional assays demonstrated that the
nonsense mutation led to a loss-of-function effect. Qian et
al (54) scanned TBX20
in 96 unrelated DCM patients and identified three mutations (p.L196
V, p.R334Q and p.W349R) in three patients, respectively. However,
the functional effect of these DCM-associated mutations remains to
be elucidated. Zhao et al (55) screened TBX20 in 120
unrelated patients with idiopathic DCM and identified a mutation
(p.F256I) in one patient. Functional analyses indicated that the
mutation had a dominant-negative effect. In the present study, a
novel TBX20 mutation that caused haploinefficiency was
associated with isolated DCM in humans for, to the best of our
knowledge, the first time. These results suggest that TBX20
mutations are an uncommon cause for DCM.
The association of the TBX20 loss-of-function
mutation with increased susceptibility to DCM may be partially
ascribed to the abnormal development and structural remodeling of
the heart. In mice, as a transcriptional regulator required for
cardiac development, TBx20 served a crucial role in the
maintenance of structural and functional phenotypes in adult mouse
heart, and homozygous deletion of TBX20 led to embryonic
lethality resulting from an underdeveloped heart that was poorly
proliferative and lacked myocardial chambers; while adult
heterozygous TBx20-null mice presented with DCM and mild ASD
(53). In addition, conditional
knockout of TBx20 in adult murine cardiomyocytes resulted in
a rapid onset and progression of heart failure, arrhythmias and
death (56). In humans and mice
during heart failure, TBX20 expression has been reported to be
downregulated, accompanied by elevated cardiomyocyte apoptotic
levels (57). In cultured neonatal
rat cardiomyocytes, H2O2 was observed to
result in a concurrent reduction in TBX20 expression and increase
in apoptosis, whereas TBX20 overexpression reduced
H2O2-induced cardiomyocyte apoptosis. In
addition, estrogen was able to protect cardiomyocytes from
H2O2-induced apoptosis by upregulating TBX20
expression in a concentration-dependent manner; while TBX20
silencing increased oxidative stress-induced apoptosis in H9c2
cells (57). These experimental
observations provide evidence supporting the hypothesis that in
humans, genetically defective TBX20 predisposes to DCM.
Previous studies demonstrated that TBX20
transcriptionally activated multiple cardiac target genes,
including ANF, CX40 and SRF, alone or in
synergy with cooperative partners (18), and loss-of-function mutations in
several transcriptionally cooperative partners of TBX20, including
NKX2-5, GATA4, GATA5, GATA6 and TBX5, have been associated with DCM
in humans (41–49). Therefore, TBX20 mutations
may enhance vulnerability to DCM, likely by reducing synergistic
transactivation of certain target genes key to embryonic
cardiogenesis and adult cardiac structure and function.
Notably, multiple TBX20 mutations have been
previously reported to be responsible for various CHDs, including
ASD, ventricular septal defect, tetralogy of Fallot, pentalogy of
Fallot, patent ductus arteriosus, common atrioventricular canal,
double outlet right ventricle, coarctation of the aorta, total
anomalous pulmonary venous connection and cardiac valvular
malformation, with ASD being the most common phenotype (32,58–61).
In the current study, ASD was documented in two family members
carrying the TBX20 mutation, including a female adult who
additionally presented with adult-onset DCM and her juvenile
daughter had only ASD without DCM. These results highlighted the
pivotal role of TBX20 in humans during cardiovascular
development.
There were limitations to the present study.
Firstly, due to the fact that other DCM-associated genes were not
screened in this family, it was not possible to rule out the
contribution of other mutant genes to the pathogenesis of the
disease. In addition, due to lack of availability of tissue samples
from the mutation carrier, it was not possible to verify the
presence of NMD, although the occurrence of NMD yielded
haploinefficiency, an effect identical to that of the functional
characterization. Finally, more functional analyses, including
analyses of subcellular distribution and the binding ability of the
mutant TBX20 to target DNA molecules or other components in the
transcriptional machinery, are required in order to explain why the
mutant TBX20 lost transcriptional activity.
In conclusion, the current study associated
TBX20 haploinefficiency with isolated DCM, and expanded upon
the mutational spectrum of TBX20 associated with DCM and
CHD, which provides novel insight into the molecular mechanism of
DCM and CHD, suggesting potential implications for early
personalized treatment of these diseases.
Acknowledgments
The authors would like to thank the participants for
their participation in the current study. The present study was
supported by grants from the National Natural Science Foundation of
China (grant nos. 81270161 and 81470372), the Key Program for Basic
Research of Shanghai, China (grant no. 14JC1405500) and the Natural
Science Fund of Shanghai, China (grant no. 15ZR1438100).
References
|
1
|
Maron BJ, Towbin JA, Thiene G,
Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB;
American Heart Association; et al Contemporary definitions and
classification of the cardiomyopathies; An American Heart
Association Scientific Statement from the Council on Clinical
Cardiology, Heart Failure and Transplantation Committee; Quality of
Care and Outcomes Research and Functional Genomics and
Translational Biology Interdisciplinary Working Groups; Council on
Epidemiology and Prevention: Circulation. 113:1807–1816. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Garcia-Pavia P, Cobo-Marcos M,
Guzzo-Merello G, Gomez-Bueno M, Bornstein B, Lara-Pezzi E, Segovia
J and Alonso-Pulpon L: Genetics in dilated cardiomyopathy. Biomark
Med. 7:517–533. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen W, Huo J, Ma A, Bai L and Liu P: A
novel mutation of the LMNA gene in a family with dilated
cardiomyopathy, conduction system disease, and sudden cardiac death
of young females. Mol Cell Biochem. 382:307–311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Agrawal PB, Pierson CR, Joshi M, Liu X,
Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC,
Haliloğlu G, et al: SPEG interacts with myotubularin, and its
deficiency causes centronuclear myopathy with dilated
cardio-myopathy. Am J Hum Genet. 95:218–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arndt AK, Schafer S, Drenckhahn JD, Sabeh
MK, Plovie ER, Caliebe A, Klopocki E, Musso G, Werdich AA, Kalwa H,
et al: Fine mapping of the 1p36 deletion syndrome identifies
mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet.
93:67–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haas J, Frese KS, Peil B, Kloos W, Keller
A, Nietsch R, Feng Z, Müller S, Kayvanpour E, Vogel B, et al: Atlas
of the clinical genetics of human dilated cardiomyopathy. Eur Heart
J. 36:1123–1135a. 2015. View Article : Google Scholar
|
|
8
|
Reinstein E, Orvin K, Tayeb-Fligelman E,
Stiebel-Kalish H, Tzur S, Pimienta AL, Bazak L, Bengal T, Cohen L,
Gaton DD, et al: Mutations in TAX1BP3 cause dilated cardiomyopathy
with septooptic dysplasia. Hum Mutat. 36:439–442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wahbi K, Béhin A, Bécane HM, Leturcq F,
Cossée M, Laforêt P, Stojkovic T, Carlier P, Toussaint M, Gaxotte
V, et al: Dilated cardiomyopathy in patients with mutations in
anoctamin 5. Int J Cardiol. 168:76–79. 2013. View Article : Google Scholar
|
|
10
|
Flack E and Kannankeril PJ: The genetics
of dilated cardiomyopathy. Heart Rhythm. 9:397–398. 2012.
View Article : Google Scholar
|
|
11
|
Pashmforoush M, Lu JT, Chen H, Amand TS,
Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, et
al: Nkx2-5 pathways and congenital heart disease; loss of
ventricular myocyte lineage specification leads to progressive
cardiomyopathy and complete heart block. Cell. 117:373–386. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prall OW, Menon MK, Solloway MJ, Watanabe
Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet
H, et al: An Nkx2-5/Bmp2/Smad1 negative feedback loop controls
heart progenitor specification and proliferation. Cell.
128:947–959. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Greulich F, Rudat C and Kispert A:
Mechanisms of T-box gene function in the developing heart.
Cardiovasc Res. 91:212–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Oka T, Xu J and Molkentin JD:
Re-employment of developmental transcription factors in adult heart
disease. Semin Cell Dev Biol. 18:117–131. 2007. View Article : Google Scholar
|
|
16
|
Song K, Nam YJ, Luo X, Qi X, Tan W, Huang
GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al: Heart
repair by reprogramming non-myocytes with cardiac transcription
factors. Nature. 485:599–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stennard FA, Costa MW, Elliott DA, Rankin
S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau
BG, et al: Cardiac T-box factor Tbx20 directly interacts with
Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the
developing heart. Dev Biol. 262:206–224. 2003. View Article : Google Scholar
|
|
19
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al: A novel NKX2.5
loss-of-function mutation associated with congenital bicuspid
aortic valve. Am J Cardiol. 114:1891–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, et al: GATA4 mutations cause human
congenital heart defects and reveal an interaction with TBX5.
Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, et al: GATA4
loss-of-function mutations underlie familial tetralogy of Fallot.
Hum Mutat. 34:1662–1671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of fallot. Int J Med Sci. 10:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar
|
|
26
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, et al: GATA5 loss-of-function
mutations associated with congenital bicuspid aortic valve. Int J
Mol Med. 33:1219–1226. 2014.PubMed/NCBI
|
|
27
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
28
|
Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R,
Chen S and Sun K: Identification of two novel GATA6 mutations in
patients with nonsyndromic conotruncal heart defects. Mol Med Rep.
10:743–748. 2014.PubMed/NCBI
|
|
29
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic tetralogy of
Fallot. Int J Mol Med. 31:51–58. 2013.
|
|
30
|
Baban A, Postma AV, Marini M, Trocchio G,
Santilli A, Pelegrini M, Sirleto P, Lerone M, Albanese SB, Barnett
P, et al: Identification of TBX5 mutations in a series of 94
patients with tetralogy of Fallot. Am J Med Genet A.
164A:3100–3107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Al-Qattan MM and Abou Al-Shaar H:
Molecular basis of the clinical features of Holt-Oram syndrome
resulting from missense and extended protein mutations of the TBX5
gene as well as TBX5 ntragenic duplications. Gene. 560:129–136.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H,
Wang J, Zhao CM, Qiu XB, Yang YQ and Liu XY: TBX20 loss-of-function
mutation contributes to double outlet right ventricle. Int J Mol
Med. 35:1058–1066. 2015.PubMed/NCBI
|
|
33
|
Andersen TA, Troelsen Kde L and Larsen LA:
Of mice and men: Molecular genetics of congenital heart disease.
Cell Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar :
|
|
34
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: A novel NKX2.5 loss-of-function mutation responsible for
familial atrial fibrillation. Int J Mol Med. 31:1119–1126.
2013.PubMed/NCBI
|
|
35
|
Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang
WY, Liu X and Yang YQ: Prevalence and spectrum of Nkx2.5 mutations
associated with idiopathic atrial fibrillation. Clinics (Sao
Paulo). 68:777–784. 2013. View Article : Google Scholar
|
|
36
|
Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang
J, Li RG, Xu L, Jiang WF, Qiu XB, et al: Mutational spectrum of the
NKX2-5 gene in patients with lone atrial fibrillation. Int J Med
Sci. 11:554–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Perera JL, Johnson NM, Judge DP and
Crosson JE: Novel and highly lethal NKX2.5 missense mutation in a
family with sudden death and ventricular arrhythmia. Pediatr
Cardiol. 35:1206–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.
|
|
39
|
Postma AV, van de Meerakker JB, Mathijssen
IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne
Deprez RH and Moorman AF: A gain-of-function TBX5 mutation is
associated with atypical Holt-Oram syndrome and paroxysmal atrial
fibrillation. Circ Res. 102:1433–1442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hong K and Xiong Q: Genetic basis of
atrial fibrillation. Curr Opin Cardiol. 29:220–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Costa MW, Guo G, Wolstein O, Vale M,
Castro ML, Wang L, Otway R, Riek P, Cochrane N, Furtado M, et al:
Functional characterization of a novel mutation in NKX2-5
associated with congenital heart disease and adult-onset
cardiomyopathy. Circ Cardiovasc Genet. 6:238–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu
YJ, Liu X, Fang WY, Yang YQ and Liao DN: A novel NKX2-5
loss-of-function mutation predisposes to familial dilated
cardiomyopathy and arrhythmias. Int J Mol Med. 35:478–486.
2015.
|
|
43
|
Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X,
Xu YJ, Jiang WF, Jiang JQ, Liu X, et al: GATA4 loss-of-function
mutation underlies familial dilated cardiomyopathy. Biochem Biophys
Res Commun. 439:591–596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.
|
|
45
|
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG
and Yang YQ: Prevalence and spectrum of GATA4 mutations associated
with sporadic dilated cardiomyopathy. Gene. 548:174–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.
|
|
47
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al: GATA6 loss-of-function
mutations contribute to familial dilated cardiomyopathy. Int J Mol
Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
48
|
Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM,
Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al: TBX5 loss-of-function
mutation contributes to familial dilated cardiomyopathy. Biochem
Biophys Res Commun. 459:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou W, Zhao L, Jiang JQ, Jiang WF, Yang
YQ and Qiu XB: A novel TBX5 loss-of-function mutation associated
with sporadic dilated cardiomyopathy. Int J Mol Med. 36:282–288.
2015.PubMed/NCBI
|
|
50
|
Akazawa H and Komuro I: Cardiac
transcription factor Csx/Nkx2-5: Its role in cardiac development
and diseases. Pharmacol Ther. 107:252–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kervestin S and Jacobson A: NMD: A
multifaceted response to premature translational termination. Nat
Rev Mol Cell Biol. 13:700–712. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Keeling KM and Bedwell DM: Suppression of
nonsense mutations as a therapeutic approach to treat genetic
diseases. Wiley Interdiscip Rev RNA. 2:837–852. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kirk EP, Sunde M, Costa MW, Rankin SA,
Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, et al:
Mutations in cardiac T-box factor gene TBX20 are associated with
diverse cardiac pathologies, including defects of septation and
valvulogenesis and cardiomyopathy. Am J Hum Genet. 81:280–291.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Qian L, Mohapatra B, Akasaka T, Liu J,
Ocorr K, Towbin JA and Bodmer R: Transcription factor
neuromancer/TBX20 is required for cardiac function in Drosophila
with implications for human heart disease. Proc Natl Acad Sci USA.
105:19833–19838. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao CM, Bing-Sun, Song HM, Wang J, Xu WJ,
Jiang JF, Qiu XB, Yuan F, Xu JH and Yang YQ: TBX20 loss-of-function
mutation associated with familial dilated cardiomyopathy. Clin Chem
Lab Med. 54:325–332. 2016. View Article : Google Scholar
|
|
56
|
Shen T, Aneas I, Sakabe N, Dirschinger RJ,
Wang G, Smemo S, Westlund JM, Cheng H, Dalton N, Gu Y, et al: Tbx20
regulates a genetic program essential to adult mouse cardiomyocyte
function. J Clin Invest. 121:4640–4654. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shen T, Yang C, Ding L, Zhu Y, Ruan Y,
Cheng H, Qin W, Huang X, Zhang H, Man Y, et al: Tbx20 functions as
an important regulator of estrogen-mediated cardiomyocyte
protection during oxidative stress. Int J Cardiol. 168:3704–3714.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu C, Shen A, Li X, Jiao W, Zhang X and
Li Z: T-box transcription factor TBX20 mutations in Chinese
patients with congenital heart disease. Eur J Med Genet.
51:580–587. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Posch MG, Gramlich M, Sunde M, Schmitt KR,
Lee SH, Richter S, Kersten A, Perrot A, Panek AN, Al Khatib IH, et
al: A gain-of-function TBX20 mutation causes congenital atrial
septal defects, patent foramen ovale and cardiac valve defects. J
Med Genet. 47:230–235. 2010. View Article : Google Scholar :
|
|
60
|
Liu JJ, Fan LL, Chen JL, Tan ZP and Yang
YF: A novel variant in TBX20 (p.D176N) identified by whole-exome
sequencing in combination with a congenital heart disease related
gene filter is associated with familial atrial septal defect. J
Zhejiang Univ Sci B. 15:830–837. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Monroy-Muñoz IE, Pérez-Hernández N,
Rodríguez-Pérez JM, Muñoz-Medina JE, Angeles-Martínez J,
García-Trejo JJ, Morales-Ríos E, Massó F, Sandoval-Jones JP,
Cervantes-Salazar J, et al: Novel mutations in the transcriptional
activator domain of the human TBX20 in patients with atrial septal
defect. Biomed Res Int. 2015:7187862015. View Article : Google Scholar : PubMed/NCBI
|