Introduction
Glycogen storage disease-type I (GSD-I), first
described by von Gierke in 1929 (1), is an inherited deficiency of the
glucose-6-phosphatase (G6Pase) complex. G6Pase is composed
primarily of glucose-6-phosphatase-α (G6PC) and glucose-6-phosphate
translocase (G6PT) (2).
Abnormalities in G6PC and G6PT result in GSD-Ia and GSD-Ib,
respectively, which are the primary subtypes of GSD-I (2).
G6PC is a glycoprotein anchored in the endoplasmic
reticulum membrane by nine transmembrane helices. G6PC is vital for
the maintenance of glucose homeostasis as it catalyzes the final
step in the glycogenolytic and glyconeogenic pathways (2,3), and
is therefore present in numerous glucose-generating organs,
including the liver, kidney and small intestine (4,5).
G6PC deficiency or abnormality may result in GSD-Ia (2,6).
GSD-Ia is characterized by hypoglycemia, growth retardation, lactic
acidosis, high levels of lipids, hepatomegaly and nephromegaly. Due
to these clinical manifestations, patients with GSD-Ia are
typically diagnosed in childhood, and the diagnosis may be
confirmed using gene sequencing to confirm the presence of
mutations in G6PC (6,7).
G6PT, the protein involved in GSD-1b, is a transporter that
functions to facilitate glycogen entry into the cell. Similar to
GSD-Ia, GSD-Ib is characterized by hypoglycemia, high levels of
lipids and hepatomegaly. However, patients with GSD-Ib may
additionally suffer from impaired neutrophil function and
neutropenia, resulting in recurrent bacterial infections (2).
GSD-Ia is responsible for >80% of GSD-I cases
(4). The overall incidence of
GSD-Ia in the population is reported to be 1 in 100,000, with a
carrier frequency of ~1 in 150 (2). To date, 86 disease-causing mutations
in G6PC have been identified (6), including missense mutations, nonsense
mutations, frameshifts resulting from insertions and deletions, and
rare gene rearrangements (6), with
missense mutations being the most common (6). Although GSD-Ia is a pan-ethnic
disease, the incidence of specific mutations varies between ethnic
populations (4). For example, the
c.648G>T mutation is prevalent in East Asians (4), whereas c.247C>T is common in the
Jewish population (6). G6PC
mutations may be classified into three categories based on their
predicted topological position: Non-helical, helical and active
site. Typically, non-helical mutations have a lesser effect on
enzymatic activity compared with helical mutations (8), and in vitro experiments have
demonstrated that G6PC containing non-helical mutations may retain
enzymatic activity (9). However,
the precise association between mutations and disease severity
remains to be fully elucidated. In the present study, a missense
mutation of G6PC was identified in a 3-month-old female
patient from a consanguineous family. The association between the
G6PC genotype and disease phenotype in this patient is in
contrast to that previously described (8). The results of the present study
suggested that the prevailing view that mutations in non-helical
regions of G6PC result only in mild phenotypic
manifestations may require revision.
Materials and methods
Patient characteristics
The 3-month-old female patient with hypoglycemia,
hepatocelluar dysfunction and lactic acidosis was groaning
frequently. The patient was in a clear state of mind and not
convulsing. The parents brought her to The Second Affiliated
Hospital of Wenzhou Medical University (Wenzhou, China), for an
emergency visit, following which she was admitted to the Department
of Pediatrics. She weighed 6 kg and was 60 cm. The patient did not
suffer from severe recurrent infections. Her parents are first
cousins (Fig. 1A). There was no
history of note. A comprehensive examination of the patient was
performed, including a physical examination, vein blood gas
analysis, an electrolyte test, an abdominal sonography and
biochemical analyses.
Molecular screening
Informed consent was obtained from everyone involved
in the present study. The study protocol was approved by The Second
Affiliated Hospital of Wenzhou Medical University Ethics Committee
(Wenzhou, China). Genomic DNA was isolated from peripheral blood
samples using a genomic DNA extraction kit (RelaxGene Blood DNA
system; Tiangen Biotech Co., Ltd., Beijing, China), according to
the manufacturer's protocols. The five exons and flanking introns
of the G6PC gene were amplified by polymerase chain reaction
(PCR) with forward, AGTCCACTTCGAACAGCCAG and reverse,
CCGATCGCGTCATGGAAAAC primers. The 50 µl total reaction
volume contained 0.4 µmol/l of each primer, 0.2 mmol/l
dNTPs, 1X PCR buffer, 50 ng genomic DNA, and 1 unit Taq DNA
polymerase (Takara Biotechnology Co., Ltd., Dalian, China). The PCR
cycling conditions consisted of denaturation at 95°C for 4 min
followed by 40 thermal cycles of 95°C for 30 sec, 58°C for 30 sec,
and 72°C for 42 sec. Resulting PCR products were electrophoresed on
a 1.5% agarose gel and purified using a PCR purification kit
(AxyPrep PCR Cleanup kit; Hangzhou Axygen Biotechnology Ltd.,
Hangzhou, China). The PCR products were sequenced using an
automated sequencer (ABI Prism 3100 Genetic Analyzer; Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, USA), and
compared with a reference sequence. The gene sequences surrounding
the mutation site in various species were obtained from the
National Center for Biotechnology Information (www.ncbi.nlm.nih.gov/pubmed/) and analyzed to
determine whether the mutation site was conserved.
Results
Clinical examination
The 3-month-old female patient was born in a
consanguineous family (Fig. 1A).
She was groaning frequently. There was no history of note. Physical
examination revealed shortness of breath, symmetrical rough breath
sounds, regular respiratory rhythm, and abdominal distension with
enlarged liver (7 cm below the right costal margin). The spleen was
not palpable. An increased level of lactic acid revealed that the
patient had lactic acidosis. Vein blood gas analysis revealed that
pH, PCO2 and HCO3− levels were
below normal, and that the absolute value of base excess was
increased, suggesting metabolic acidosis (Table I). In addition, the shortness of
breath exhibited by the patient indicated metabolic acidosis. A
fasting blood glucose level of 2.73 mmol/l indicated that the
infant suffered from severe hypoglycemia. Furthermore, increased
levels of alanine aminotransferase, aspartate aminotransferase, and
γ-glutamyl transpeptidase suggested liver dysfunction.
Additionally, the levels of uric acid and triglycerides were also
increased (Table II). Abdominal
sonography identified hepatomegaly with enhanced echo texture
(Fig. 1B) and nephromegaly
(Fig. 1C), with the size of left
kidney measured at 72×40 mm and the size of right kidney measured
at 71×30 mm. The enlarged organs indicated the severity of the
disease. A viral hepatitis serology screen revealed that the
patient was positive for hepatitis B surface antigen (461.5),
negative for hepatitis B core antibody, and that total
immunoglobulin was normal. The patient was negative for human
immunodeficiency virus. In summary, the patient's clinical
manifestations and test results suggested GSD-Ia.
 | Table IResults of patient vein blood gas
analysis. |
Table I
Results of patient vein blood gas
analysis.
| Parameter | Test value | Change | Reference range |
|---|
| pH | 7.26 | ↓ | 7.35–7.45 |
| PCO2,
mmHg | 24.0 | ↓ | 35–45 |
| HCO3,
mmol/l | 10.3 | ↓ | 22–27 |
| BE, mmol/l | −15.1 | ↑ | −2–3 |
| Table IIResults of patient blood biochemical
analysis. |
Table II
Results of patient blood biochemical
analysis.
| Parameter | Test value | Change | Reference range |
|---|
| FBG, mmol/l | 2.73 | ↓ | 3.90–6.10 |
| Lactic acid,
mmol/l | 12.20 | ↑ | 0.6–2.20 |
| ALT, U/l | 102 | ↑ | 7–40 |
| AST, U/l | 169 | ↑ | 13–35 |
| GGT, U/l | 820 | ↑ | 7–38 |
| UA,
µmol/l | 1035 | ↑ | 89–357 |
| TG, mmol/l | 7.90 | ↑ | 0.56–1.70 |
Diagnosis of GSD-Ia
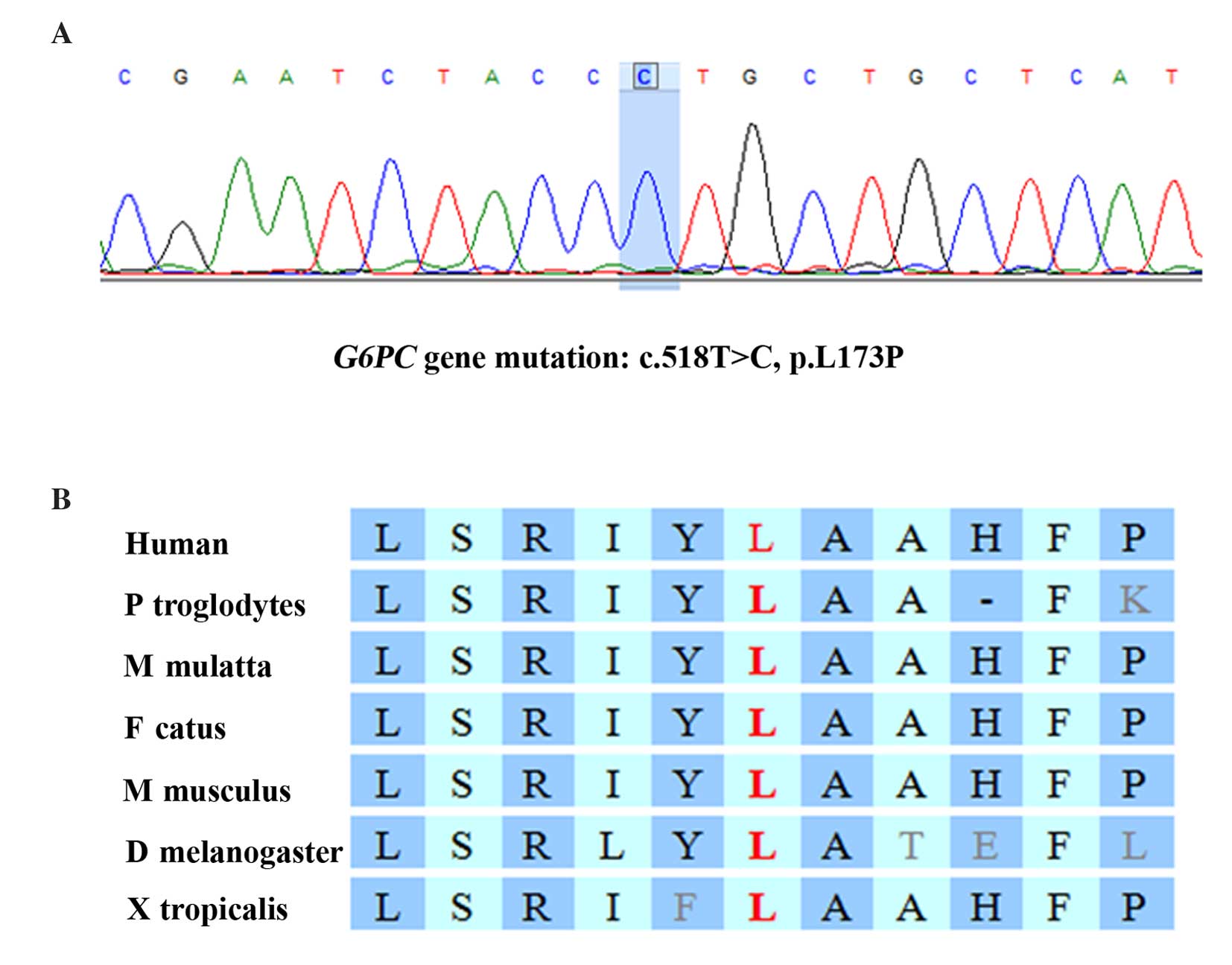
Gene sequencing revealed that the patient was
homozygous for a missense mutation in the G6PC gene,
c.518T>C (p.L173P; Fig. 2A),
which changed the codon for leucine at position 518 to a codon for
proline. Pedigree analysis revealed that the patient's father and
mother are heterozygous for this mutation (Fig. 1A). By comparing the G6PC
sequence across various species, it was determined that the
mutation is located in a highly conserved region of the G6PC
gene (Fig. 2B). Furthermore, the
p.L173P mutation is absent from the 1000G database (http://www.1000genomes.org/), suggesting it is a very
rare mutation. This finding, together with the severe symptoms of
the patient, suggested that there is a high probability that the
p.L173P mutation results in abnormal G6PC enzyme function. However,
this mutation affects a non-helical region of the protein, which
previous studies suggest are of less importance compared with the
helical regions for enzyme function (8). Therefore, the phenotypic
characteristics associated with a mutation in the non-helical
region of the protein are expected to be mild. However, in the
present study the clinical manifestations were severe.
Discussion
G6PC is a 357-amino acid glycoprotein anchored to
the ER membrane by nine transmembrane helices, with an enzymatic
active site facing the ER lumen (8). The amino-terminus of G6PC is located
in the ER lumen, and the carboxyl-terminus is in the cytoplasm
(4,8,10).
The G6PC gene, located on chromosome 17q21, is a single copy
gene composed of five exons (10).
The only known cause of GSD-Ia is mutation of G6PC.
Mutations have been divided into three categories, non-helical,
helical and active site, based on their predicted topological
position. In general, non-helical mutations are thought to have a
smaller effect on enzyme activity compared with those in
transmembrane helices (8). In the
present study, the case of a 3-month-old infant with a c.518T>C
(p.L173P) non-helical G6PC mutation is reported. According
to the prevailing view, the infant would be expected to have mild
phenotypic characteristics. However, the clinical manifestations
were clear, as the patient suffered from severe hypoglycemia and
hepatomegaly. Abdominal sonography revealed nephromegaly. Li et
al (11) reported a 3-year-old
girl who was not diagnosed with GSD-Ia until the age of 2. The
phenotypic characteristics of this patient were fasting
hypoglycemia, hepatomegaly, hypercholesterolemia and recurrent
epistaxis. The patient did not suffer from nephromegaly or
metabolic acidosis. Her biochemical results were improved through a
diet of regular uncooked cornstarch and frequent carbohydrate
feeds. That patient was not born of a consanguineous union. DNA
sequencing identified that the mother had a heterozygous R83H
(G327A) mutation and the father had a heterozygous L173P (T597C)
mutation (11). Thus, the patient
had a p.L173P mutation, as in the present study. While the two
girls suffered from identical mutations, the severity of their
phenotypic characteristics was very different. The results of the
present study suggested that non-helical mutations may result in
severe clinical manifestations, and that phenotypic heterogeneity
exists in p.L173P G6PC mutations.
GSD-Ia is a pan-ethnic disorder; however, the
incidence of mutations varies between ethnicities. To date, no
reports of a p.L173P mutation have been presented in any ethnicity
other than Chinese. This may indicate that the incidence of the
p.L173P mutation is relatively high in the Chinese population.
In the present study, hypoglycemia and lactic
acidosis were under control following administration of intravenous
glucose to the infant at a dose of 6 mg/kg/min. Nocturnal
nasogastric infusion of glucose and intermittent oral
administration of uncooked cornstarch are the primary dietary
therapies for GSD-Ia (12).
However, even with dietary therapy, long-term clinical problems may
occur. These include growth retardation, hepatomegaly, intermittent
hypoglycemia, lactic acidemia, hyperlipidemia, adenoma, gout
associated with hyperuricemia, proteinuria and nephrolithiasis with
progressive renal failure. Replacement therapy is a promising
alternative strategy for GSD-Ia treatment, and may involve
administration of G6PC, liver-kidney transplantation or gene
therapy. As G6PC is hydrophobic, it is difficult to isolate in an
active, soluble form that would facilitate its administration to
patients (5). Case reports have
revealed that liver-kidney transplantation may rectify the
metabolic disorder associated with GSD-Ia (13,14).
However, liver-kidney transplantation brings with it associated
risks and challenges. Gene therapy, while not yet developed for the
clinic, may in the future provide a strategy for the treatment of
GSD-Ia (15,16). Previous studies have demonstrated
the efficacy of gene therapy in treating mouse models of GSD-Ia
(15,17,18).
In addition, alternative therapies, including bone marrow-derived
myelomonocyte transplantation, may have the potential to restore
normal liver function (19).
In conclusion, the present study reports the case of
a 3-month-old female Chinese patient with GSD-Ia and identified a
G6PC missense mutation of c.518T>C (p.L173P) located in a
highly conserved area. The severity of the phenotype was
inconsistent with that predicted by her genotype. The incidence of
the p.L173P mutation may be relatively high in the Chinese
population. Knowledge of the various phenotypic presentations of
the p.L173P mutation may be beneficial for future
investigations.
Acknowledgments
The authors appreciate the participation of the
patient and her family in the present study. The present study was
supported by the Zhejiang Provincial Natural Science Foundation of
China (no. Y2100530, awarded to X.S.), the Wenzhou Science and
Technology Foundation (no. Y20140358, awarded to X-F.H.) and the
Key Scientific and Technological Innovation Team of Wenzhou (no.
C20150004, awarded to Z-B.J.).
References
|
1
|
Moses SW: Historical highlights and
unsolved problems in glycogen storage disease type 1. Eur J
Pediatr. 161(Suppl 1): S2–S9. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koeberl DD, Kishnani PS, Bali D and Chen
YT: Emerging therapies for glycogen storage disease type I. Trends
Endocrinol Metab. 20:252–258. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hutton JC and O'Brien RM:
Glucose-6-phosphatase catalytic subunit gene family. J Biol Chem.
284:29241–29245. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gu LL, Li XH, Han Y, Zhang DH, Gong QM and
Zhang XX: A novel homozygous no-stop mutation in G6PC gene from a
Chinese patient with glycogen storage disease type Ia. Gene.
536:362–365. 2014. View Article : Google Scholar
|
|
5
|
Resaz R, Vanni C, Segalerba D, Sementa AR,
Mastracci L, Grillo F, Murgia D, Bosco MC, Chou JY, Barbieri O, et
al: Development of hepatocellular adenomas and carcinomas in mice
with liver-specific G6Pase-α deficiency. Dis Model Mech.
7:1083–1091. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kishnani PS, Austin SL, Abdenur JE, Arn P,
Bali DS, Boney A, Chung WK, Dagli AI, Dale D, Koeberl D, et al:
Diagnosis and management of glycogen storage disease type I: A
practice guideline of the American college of medical genetics and
genomics. Genet Med. 16:e12014.PubMed/NCBI
|
|
7
|
Wang J, Cui H, Lee NC, Hwu WL, Chien YH,
Craigen WJ, Wong LJ and Zhang VW: Clinical application of massively
parallel sequencing in the molecular diagnosis of glycogen storage
diseases of genetically heterogeneous origin. Genet Med.
15:106–114. 2013. View Article : Google Scholar
|
|
8
|
Chou JY and Mansfield BC: Mutations in the
glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen
storage disease. Hum Mutat. 29:921–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shieh JJ, Terzioglu M, Hiraiwa H, Marsh J,
Pan CJ, Chen LY and Chou JY: The molecular basis of glycogen
storage disease type 1a: Structure and function analysis of
mutations in glucose-6-phosphatase. J Biol Chem. 277:5047–5053.
2002. View Article : Google Scholar
|
|
10
|
Chou JY, Jun HS and Mansfield BC: Glycogen
storage disease type I and G6Pase-β deficiency: Etiology and
therapy. Nat Rev Endocrinol. 6:676–688. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li DZ, Liao C and Tang XW: Prenatal
diagnosis of glycogen storage disease type Ia, presenting a new
mutation in the glucose-6-phosphatase gene. Prenat Diagn.
27:685–686. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shah KK and O'Dell SD: Effect of dietary
interventions in the maintenance of normoglycaemia in glycogen
storage disease type 1a: A systematic review and meta-analysis. J
Hum Nutr Diet. 26:329–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Belingheri M, Ghio L, Sala A, Menni F,
Trespidi L, Ferraresso M, Berardinelli L, Rossi G, Edefonti A and
Parini R: Combined liver-kidney transplantation in glycogen storage
disease Ia: A case beyond the guidelines. Liver Transpl.
13:762–764. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marega A, Fregonese C, Tulissi P, Vallone
C, Gropuzzo M, Toniutto PL, Baccarani U, Bresadola F, Toso F and
Montanaro D: Preemptive liver-kidney transplantation in von Gierke
disease: A case report. Transplant Proc. 43:1196–1197. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yiu WH, Lee YM, Peng WT, Pan CJ, Mead PA,
Mansfield BC and Chou JY: Complete normalization of hepatic G6PC
deficiency in murine glycogen storage disease type Ia using gene
therapy. Mol Ther. 18:1076–1084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee YM, Jun HS, Pan CJ, Lin SR, Wilson LH,
Mansfield BC and Chou JY: Prevention of hepatocellular adenoma and
correction of metabolic abnormalities in murine glycogen storage
disease type Ia by gene therapy. Hepatology. 56:1719–1729. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee YM, Pan CJ, Koeberl DD, Mansfield BC
and Chou JY: The upstream enhancer elements of the G6PC promoter
are critical for optimal G6PC expression in murine glycogen storage
disease type Ia. Mol Genet Metab. 110:275–280. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Clar J, Mutel E, Gri B, Creneguy A,
Stefanutti A, Gaillard S, Ferry N, Beuf O, Mithieux G, Nguyen TH
and Rajas F: Hepatic lentiviral gene transfer prevents the
long-term onset of hepatic tumours of glycogen storage disease type
1a in mice. Hum Mol Genet. 24:2287–2296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Resaz R, Emionite L, Vanni C, Astigiano S,
Puppo M, Lavieri R, Segalerba D, Pezzolo A, Bosco MC, Oberto A, et
al: Treatment of newborn G6pc(-/-) mice with bone marrow-derived
myelomonocytes induces liver repair. J Hepatol. 55:1263–1271. 2011.
View Article : Google Scholar : PubMed/NCBI
|