Introduction
Obesity is considered to be a systemic, chronic
low-grade inflammation, characterized by increased serum levels of
pro-inflammatory proteins and accumulation of macrophages within
white adipose tissue (1). Adipose
tissues secrete a variety of proinflammatory mediators, including
tumor necrosis factor-α (TNF-α), monocyte chemoattractant protein-1
(MCP-1) and keratinocyte-derived chemokine (KC). MCP-1 and KC are
involved in driving macrophage accumulation and activation, which
are important steps towards establishing inflammation in adipose
tissues (2). Acylation stimulating
protein (ASP) is an adipokine, which is produced by adipocytes and
interacts with C5aR-like receptor 2, a seven transmembrane G
protein-coupled receptor. In humans, circulating levels of ASP are
increased in obesity, and in insulin resistance, diabetes,
cardiovascular diseases and metabolic syndrome, even in the absence
of obesity. By contrast, the levels of ASP decrease with weight
loss or exercise (3). These
previous findings suggest the possibility that ASP is involved in
the regulation of adipose tissue inflammation. Nicotine, a
selective cholinergic agonist, is involved in cholinergic
anti-inflammatory activities in vitro and in vivo, by
acting through the α7 nicotinic acetylcholine receptor (α7nAChR).
The anti-inflammatory activity of α7nAChR has been demonstrated in
various disease models, including arthritis, septic shock and
endotoxemia (4–6). Previous studies have reported that an
α7nAChR-selective agonist, TC-7020, reduces food intake and weight
gain, levels of circulating glucose and triglycerides, and
expression levels of proinflammatory cytokines. These effects are
reversed by the α7nAChR antagonist, methyllycaconitine, supporting
the involvement of α7nAChR (7). In
addition, the expression of α7nAChR is downregulated in obese
adults, compared with adults of a healthy weight, and weight loss
has been found to partially restore the expression of α7nAChR
(8). These studies indicate the
possibility that α7nAChR affects the inflammation of adipose
tissues and may be a promising target for therapies aimed at
obesity-associated inflammatory diseases.
Nuclear factor-κB (NF-κB) is a ubiquitous, rapid
response transcription factor, which is involved in inflammatory
reactions, and exerts its actions by translating several cytokines,
chemokines and cell adhesion molecules. Several reviews have
provided evidence that NF-κB inflammatory pathways promote
metabolic diseases, including insulin resistance and
atherosclerosis (9–11), and several studies have shown that
p38 kinase is a key member of the mitogen-activated protein kinase
(MAPK) family, which is in adipocyte differentiation and
adipogenesis, and in regulating cell proliferation, inflammation
and immune responses (12–17). The anti-inflammatory actions of
α7nAChR are mediated by the inhibition of NF-κB and p38 kinase in
several types of cells, including monocytes, macrophages and
endothelial cells (17–20). However, the specific role of p38
kinase and NF-κB in ASP signaling nd the possible molecular
mechanism underlying the intracellular signal transduction from
α7nAChR leading to the anti-inflammatory action in adipocytes
remain to be fully elucidated.
In the present study, the involvement of α7nAChR on
ASP-induced cytokine production, and its mechanisms of action, were
investigated. It was found that the activation of α7nAChR in 3T3-L1
adipocyte cells inhibited the ASP-induced production of MCP-1 and
KC by inhibiting the ASP-induced activation of p38 kinase and
NF-κB.
Materials and methods
Materials and reagents
Media and anti-KC polyclonal antibody (cat. no.
PA1-32924) were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Monocyte chemoattractant protein-1 (MCP-1; cat.
no. sc-28879), inhibitor of NF-κB (IκB)α (cat. no. sc-847), NF-κB
(cat. no. sc-109) and poly (ADP-ribose) polymerase (PARP; cat. no.
sc-8007) antibodies were purchased from Santa Cruz Biotechnology,
Inc (Santa Cruz, CA, USA). Antibodies against phosphorylated-p38
kinase (cat. no. 9211) and p38 kinase (cat. no. 9212; Cell
Signaling Technology, Inc., Beverly, MA, USA) were used to detect
the phosphorylated form of p38 kinase and total p38 kinase,
respectively. Anti-β-actin antibody (cat. no. SAB5500001),
horseradish peroxidase-labeled goat anti-mouse IgG and the NF-κB
inhibitor, BAY-11-7082, were purchased from Sigma-Aldrich (St.
Louis, MO, USA). The 3T3-L1 preadipocytes were purchased from
American Type Culture Collection (Manassas, VA, USA), and the
ProteoExtract™ subcellular proteome extraction kit was purchased
from Calbiochem; EMD Millipore (Billerica, MA, USA). Other reagents
and laboratory supplies were obtained from Sigma-Aldrich (St.
Louis, MO, USA).
Cell culture
The mouse 3T3-L1 preadipocytes were seeded at a
density of 1.0×105 cells/well in 5-well plates and
routinely cultured at 37°C in a humidified atmosphere of 5%
CO2 in air. The cells were maintained in Dulbecco's
modified Eagle's medium and Ham's F-12 nutrient mixture (DMEM/F-12)
with 1% penicillin-streptomycin (100 U/ml; 100 µg/ml) and
10% fetal bovine serum (FBS). At confluence, adipocyte
differentiation was induced by adding 1 µM dexamethasone,
0.5 mM isobutylmethylxanthine and 1 mg/l insulin for 2 days,
followed by 2 days in differentiating medium, containing 10% FBS in
DMEM/F12 with 1 mg/l insulin supplementation, and replaced every 2
days. After 8–9 days, the cells exhibited a differentiated
morphology (>80% of the cells) with lipid accumulation. Two days
post-differentiation, cells were treated with ASP or PBS control
for indicated time at 37°C. Alternatively, cells were pretreated
with nicotine (10 µM), α7nAChR antagonist α-BTX (2
µM), p38 kinase inhibitor SB203580 (20 μM), NF-κB inhibitor
BAY-11-7082 (5 µM) for 30 min, followed by stimulation with
or without ASP (100 nM) for an additional 24 h.
Recombinant ASP
Recombinant human ASP was produced and purified, as
described previously (21). To
avoid the inactivation of ASP, no denaturing agents were used at
any step in the purification process. The purity was assessed using
mass spectrometry, and the ASP was confirmed as endotoxin-free.
Preparation of cytoplasmic and nuclear
protein fractions and immunoblotting
The 3T3-L1 adipocytes were processed using a
ProteoExtract subcellular proteome extraction kit (Calbiochem; EMD
Millipore), according to the manufacturer's protocol, to produce
cytoplasmic and nuclear protein fractions, which were then analyzed
by immunoblotting using the indicated antibodies. PARP and β-actin
were used as loading controls of the nuclear and cytoplasmic
fractions, respectively (22).
Immunoblotting
The 3T3-L1 adipocytes were lysed in SDS sample
buffer, sonicated and centrifuged at 12,000 × g for 15 min at 4°C.
The resulting supernatants were boiled for 5 min in the presence of
50 mmol/l dithiothreitol. To measure the levels of secreted
proteins (MCP-1 and KC), the cultured medium of the cells was also
boiled for 5 min in SDS sample buffer (23). The fractions were sonicated and
clarified by centrifugation 12,000 × g for 15 min at 4°C, and their
protein concentrations were assessed using a BCA Protein Assay kit
(Pierce; Thermo Fisher Scientific, Inc.). Equivalent quantities of
protein were separated using 7.5~15% (depending on their molecular
weight) SDS-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes, blocked for 1 h in phosphate-buffered
saline containing 5% nonfat dry milk and 0.1% Tween 20, and
subsequently incubated with primary antibodies at 4°C overnight.
The antibodies were as follows: MCP-1 (1:200), KC (1:500), β-actin
(1:1,000), NF-κB p65 (1:500), IκBα (1:500), p38 kinase (1:1,000),
phosphorylated p38 kinase (1:500) and PARP (1:500). Following
incubation with secondary antibodies (1:5,000) at room temperature
with agitation for 1 h, the membranes were washed three times with
100 ml of Tris-buffered saline containing 1% (v/v) Tween 20. The
proteins were detected using enhanced chemiluminescence (24,25).
The PARP and β-actin signals were used for blotting to verify
equivalent gel loading. Band densities were determined using
Quantity One software version 4.62 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Statistical analysis
The results are presented as the percentage of
control values, as the mean ± standard error of the mean.
Differences between mean values of normally distributed data were
assessed by Student's t-test for single comparisons between
treatment and control. For data with multiple comparisons, one-way
analysis of variance followed by Dunnett's test were used.
P<0.05 was considered to indicate a statistically significant
difference. The data were analyzed using SPSS 17.0 (SPSS, Inc.,
Chicago, IL, USA).
Results
ASP increases the expression levels of
MCP-1 and KC
To determine the effect of ASP on the expression of
cytokines by differentiated adipocyte cultures, the present study
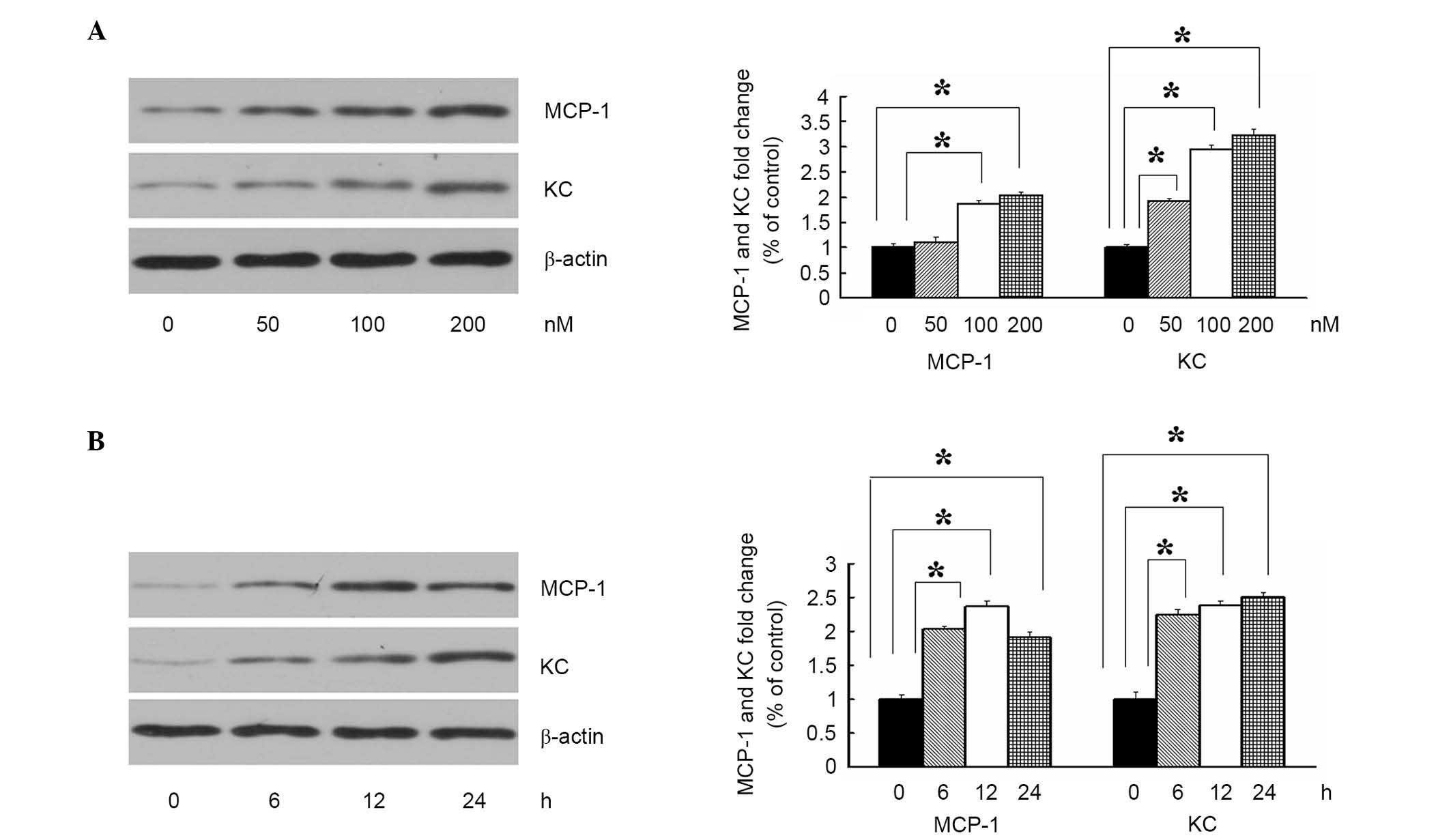
examined the levels of MCP-1 and KC using immunoblotting. As shown
in Fig. 1A, the 3T3-L1 adipocytes
were treated with increasing concentrations of ASP (0, 50, 100 and
200 nM), and the levels of MCP-1 and KC appeared to increase
gradually with increasing concentrations of ASP, compared with the
media control at 12 h, which suggested that ASP increased the
expression of cytokines in a concentration-dependent manner. The
effect of ASP on the expression of cytokines was also
time-dependent (Fig. 1B). A fixed
concentration of ASP (100 nM) increased the expression levels of
MCP-1 and KC between 6 and 4 h, reaching a maximum at 24 h. These
results indicated that ASP promoted adipocyte inflammation by
enhancing the expression of MCP-1 and KC.
α7nAChR inhibits the ASP-induced
expression of KC and MCP-1
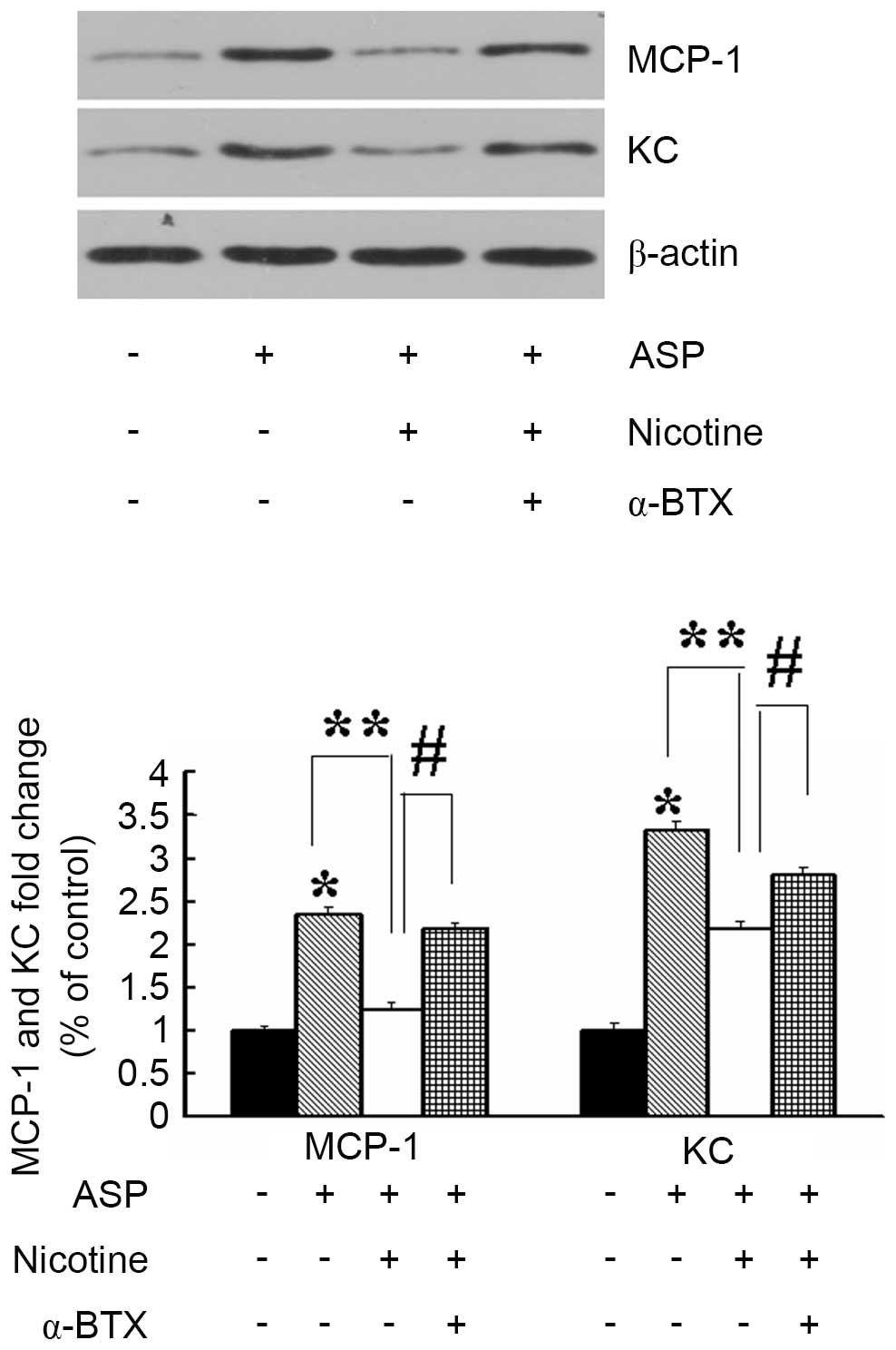
To determine the involvement of α7nAChR in the
production of proinflammatory mediators, the 3T3-L1 adipocytes were
treated with vehicle, nicotine or α-bungarotoxin (α-BTX), prior to
stimulation with ASP. Treatment of the 3T3-L1 adipocytes with ASP
(100 nM; 24 h) alone significantly induced the expression of KC and
MCP-1, compared with the control (Fig.
2). However, the production of KC and MCP-1 induced by ASP was
inhibited significantly by preincubation with nicotine (10
µM; 30 min). In addition, the suppression of cytokine
production by nicotine was prevented by the addition of α-BTX (2
µM; 30 min), which is an antagonist of α7nAChR. These
results indicated that nicotine eliminated the ASP-induced
production of inflammatory factors from adipocytes via the
stimulation of α7nAChR.
Inhibition of ASP-stimulated p38 kinase
phosphorylation by α7nAChR inhibits the expression of KC and
MCP-1
The pathways involving p38 kinase are crucial in the
regulation of pro-inflammatory molecules in cellular responses. The
present study hypothesized that nicotine inhibits the ASP-induced
production of inflammatory cytokines by interfering with the p38
kinase signaling pathway, therefore, the effect of ASP on the
activation of p38 kinase was investigated. As shown in Fig. 3A, ASP (100 nM; 24 h) resulted in a
significant increase in the phosphorylation of p38 kinase, without
affecting the overall level of total p38. Subsequently, whether
nicotine inhibited the ASP-induced production of cytokines by
modulating ASP-induced p38 kinase activation was examined. As
expected, the results of the immunoblot analysis showed that
pretreatment with 10 µM nicotine suppressed the ASP-induced
phosphorylation of p38 kinase. In addition, this was completely
reversed by α-BTX (2 µM; 30 min). These data indicated that
nicotine, through its actions on α7nAChR, inhibited the expression
of ASP-induced cytokines via the suppression of p38 kinase signal
transduction. The significance of inhibiting the ASP-induced p38
kinase activation by nicotine in the production of cytokines was
further confirmed with by p38 kinase inhibitor, SB203580 (20
µM; 30 min). As shown in Fig.
3B and C, pretreatment with SB203580 markedly inhibited the
ASP-induced production of MCP-1 and KC, and phosphorylation of p38
kinase. These data demonstrated that nicotine suppressed the
ASP-induced production of cytokines through downregulation of p38
kinase activity in the 3T3-L1 cells.
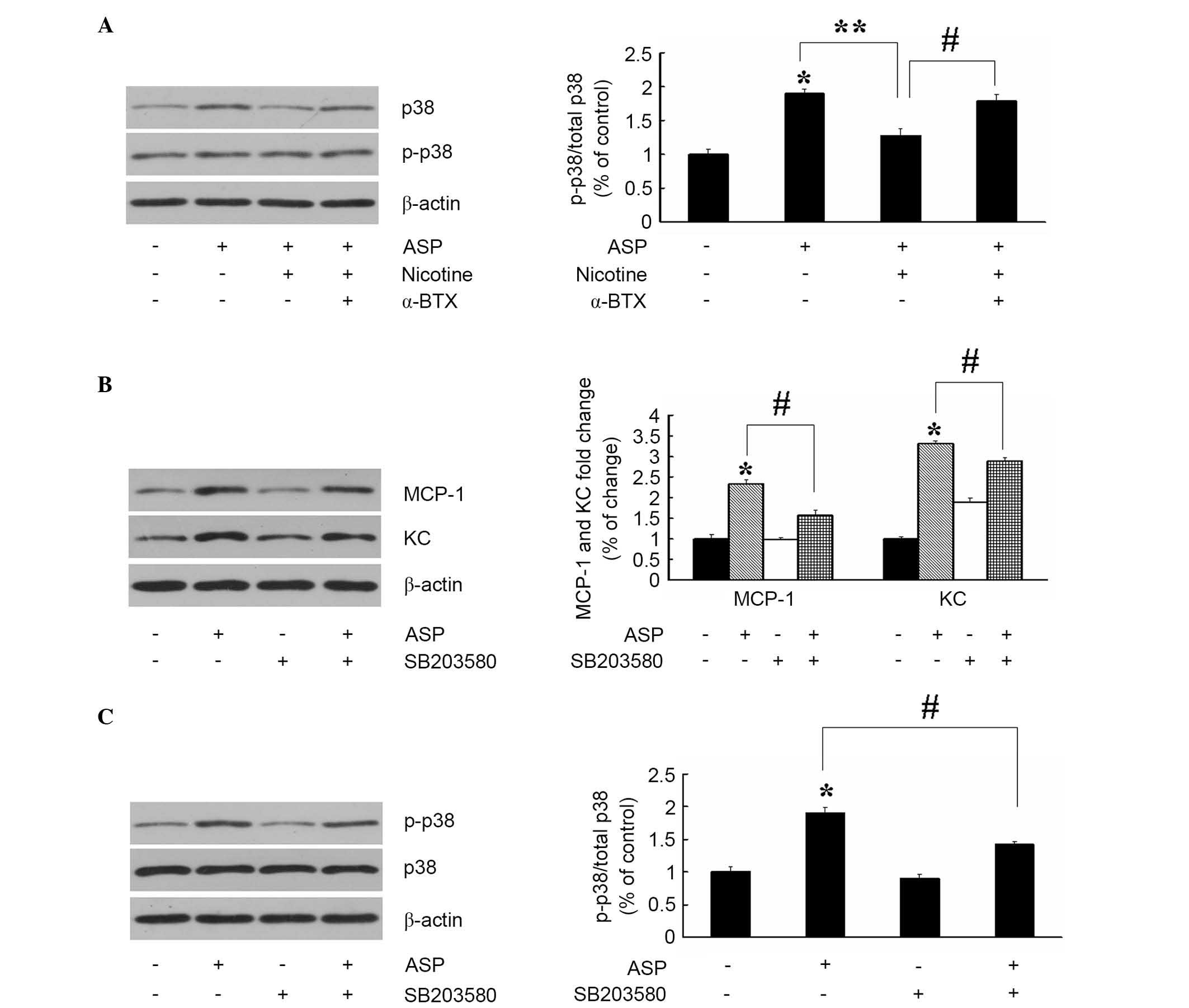 | Figure 3Inhibition of ASP-stimulated p38
kinase phosphorylation by α7nAChR inhibits the expression of KC and
MCP-1. (A) Adipocytes were pretreated with nicotine (10 µM)
in the absence or presence of α-BTX (2 µM) for 30 min and
then challenged with ASP (100 nM) for 24 h. Following treatment,
levels of p38 kinase and p-p38 kinase were determined using
immunoblot and densitometric analysis. Data are presented as the
mean ± standard error of the mean (n=3) *P<0.05, vs.
untreated control; **P<0.05, vs. ASP;
#P<0.05, vs. nicotine+ASP. Adipocytes were incubated
with ASP (100 nM) for 24 h and/or the p38 kinase inhibitor,
SB203580 (20 µM; 30 min). The levels of (B) KC and MCP-1,
and (C) p38 kinase and p-p38 kinase were determined using western
blot and densitometric analysis. Data are presented as the mean ±
standard error of the mean (n=3). *P<0.05, vs.
untreated control; #P<0.05, vs. ASP. α7nAChR, α7
nicotinic acetylcholine receptor; ASP, acylation stimulating
protein; KC, keratinocyte-derived chemokine; MCP-1, monocyte
chemoattractant protein-1; p-, phosphorylated. |
Inhibition of ASP-stimulated NF-κB
activation by α7nAChR inhibits the expression of KC and MCP-1
NF-κB is a transcription factor, which modulates the
expression of a variety of genes involved in inflammatory
responses. In an unstimulated cell, NF-κB resides in the cytoplasm
as an inactive NF-κB-IκB complex. When the cell is stimulated, IκB
becomes phosphorylated and is subsequently degraded, allowing NF-κB
to translocate into the nucleus (19,20).
As the degradation of IκB is an essential step in NF-κB activation
by various stimuli, the present study used immunoblot analysis to
examine the levels of total IκBα in the 3T3-L1 adipocytes of the
different treatment groups. As shown in Fig. 4A, the level of IκBα was markedly
decreased in the ASP (100 nM; 24 h) groups, compared with the
untreated control. Pretreatment with nicotine prevented ASP-induced
IκBα degradation. In addition, the suppression of IκBα degradation
by nicotine was prevented by the addition of α-BTX. These results
demonstrated that α7nAChR inhibited the ASP-induced production of
cytokines, partially by preventing the degradation of IκBα. As the
activation and nuclear translocation of NF-κB is an essential step
in the regulation of cytokine production, the present study
performed immunoblot analysis of the nuclear and cytosolic extracts
to assess whether nicotine altered the nuclear translocation of
NF-κB p65 induced by ASP. As shown in Fig. 4B and C, the nuclear translocation
of NF-κB p65 was induced upon treatment with ASP. However,
pretreatment with nicotine significantly attenuated the ASP-induced
translocation of NF-κB p65. In addition, the nicotinic-induced
suppression of translocation was prevented by the addition of
α-BTX. These results demonstrated that α7nAChR inhibited the
production of MCP-1 and KC in the ASP-stimulated adipocytes by
inhibiting the translocation of NF-κB p65. The present study
further confirmed the inhibition of ASP-induced NF-κB activation by
nicotine in the production of cytokines using the NF-κB inhibitor,
BAY-11-7082 (5 µM; 30 min). As shown in Fig. 4D and E, pretreatment with
BAY-11-7082 markedly inhibited the ASP-induced production of MCP-1
and KC, and inhibited the degradation of IκBα. These data
demonstrated that nicotine suppressed the ASP-induced production of
cytokines through downregulation of NF-κB activity in 3T3-L1
cells.
 | Figure 4Inhibition of ASP-stimulated NF-κB
activation by α7nAChR inhibits the expression of KC and MCP-1.
Adipocytes were pretreated with nicotine 10 µM in the
absence or presence of α-BTX (2 µM) for 30 min and then
challenged with ASP (100 nM) for 24 h. The expression levels of (A)
IκBα and the (B) p65 subunit of NF-κB in the cytosol extracts and
(C) nuclear extracts were determined using immunoblot and
densitometric analysis. Data are presented as the mean ± standard
error of the mean (n=3). *P<0.05, vs. untreated
control; **P<0.05, vs. ASP; #P<0.05,
vs. nicotine+ASP. Adipocytes were incubated with ASP (100 nM) for
24 h or the NF-κB inhibitor, BAY-11-7082 (10 nM), and the
expression levels of (D) KC and MCP-1, and (E) IκBα were determined
using immunoblot and densitometric analysis. Data are presented as
the mean ± standard error of the mean (n=3), *P<0.05,
vs. untreated control; #P<0.05, vs. ASP. α7nAChR, α7
nicotinic acetylcholine receptor; ASP, acylation stimulating
protein; α-BTX, α-bungarotoxin; NF-κB, nuclear factor-κB; IκBα,
inhibitor of NF-κB; KC, keratinocyte-derived chemokine; MCP-1,
monocyte chemoattractant protein-1; PARP, poly (ADP-ribose)
polymerase. |
Discussion
Obesity is accompanied by low-level inflammation,
and this has been regarded to be the mechanistic link between
obesity and associated cardiovascular and diabetic complications
(26). MCP-1 and KC are important
in the accumulation and activation of macrophages in inflamed
adipose tissue. In the present study, ASP increased the expression
levels of MCP-1 and KC in a concentration- and time-dependent
manner, as revealed by immunoblot assays. Consistent with these
results, Tom et al (3),
reported that ASP treatment at 200 nM increased the secretion of KC
and MCP-1, as measured using ELISA kits. These findings suggest
that ASP contributes to obesity-mediated inflammation and adipose
tissue macrophage invasion. A previous histological study showed
that ASP increases the numbers of M1 macrophages in the adipose
tissue, liver and skeletal muscle of mice (27). In addition, ASP exerts a direct
concentration-dependent effect, which increase migration and M1
activation of cultured macrophages (27).
There are at least three families of MAPKs,
including extracellular signal-regulated kinase, c-Jun-N-terminal
kinase and p38 kinase, existing in mammalian cells. As described
previously, p38 kinase is considered to be involved in the
regulation of inflammatory responses (13). In the present study, the increased
production of the ASP-stimulated cytokines, MCP-1 and KC, in the
3T3-L1 cells was partly mediated by the p38 kinase phosphorylation
pathways. The inhibition of p38 kinase following the administration
of SB203580, a selective p38 kinase inhibitor, suppressed the
ASP-induced phosphorylation of p38 kinase and production of
cytokines. The results of the present study are concordant with
those from a study on adipocytes stimulated with TNF-α, which
selectively increased the expression of MCP-1 via the activation of
p38 kinase (28).
α7nAChR is involved in mediating cholinergic
anti-inflammatory activities in vitro and in vivo. In
the present study, nicotine pretreatment significantly inhibited
the ASP-induced expression of KC and MCP-1, and this was prevented
by the α7nAChR antagonist, α-BTX. The fact that nicotine
significantly reduced the phosphorylation of p38 kinase
demonstrated that the effects of α7nAChR on the ASP-induced
production of KC and MCP-1 were mediated by inhibition of the p38
kinase pathway. The results of the present study are consistent
with previous reports that α7nAChR inhibits the
lipopolysaccharide-induced release of TNF-α in microglial cells,
and is associated with the suppression of p38 kinase activity
(29,30). By contrast, Aicher et al
(31) showed that, in dendritic
cells, nicotine induced α7nAChR-mediated T-cell proliferation and
cytokine secretion by partly activating p38 kinase. The most likely
explanation for the discrepant results is that, in these systems,
regulation is cell type- and stimulus-dependent.
NF-κB is composed of two subunits, p50 and p65, and
is retained in the cytoplasm of unstimulated cells by a
non-covalent interaction with the inhibitory molecule, IκB.
Following activation by a number of physiological and
non-physiological stimuli, IκB dissociates from NF-κB within
minutes, and undergoes ubiquitination and degradation (19,20).
Upon release, NF-κB is translocated to the nucleus, where it
regulates the transcription of inflammatory genes (32). The data in the present study showed
that treatment of the adipocytes with ASP promoted the degradation
of IκBα and translocation of NF-κB. Pretreatment with the NF-κB
inhibitor, BAY-11-7082, inhibited the ASP-induced production of
MCP-1 and KC, and degradation of IκBα. These findings are
consistent with those of a previous study, which reported that ASP
increased the phosphorylation of Ser(468) and Ser(536) of p65
NF-κB, which is required for the transactivation of gene
expression, in a time- and concentration-dependent manner (3). Pretreatment of the cells with
nicotine in the present study inhibited the ASP-induced activation
of NF-κB, and production of MCP-1 and KC. Consistent with these
results, previous studies have reported that, in different cells,
including monocytes, mast cells and endothelial cells, α7nAChR may
prevent inflammation by inhibiting NF-κB transcriptional activity
(17,19,33).
By contrast, other studies have shown that α7nAChR signaling
proceeds through intracellular pathways, leading to the upregulated
expression and transactivation of NF-κB (34,35).
These findings suggest that a different stimulus, or stimulus
intensity, targeted to the same receptor may either inhibit or
activate the same signaling system. The mechanism used by nicotine
to modulate the response of NF-κB to ASP remains to be elucidated.
The present study hypothesized that nicotine may also activate
intracellular anti-inflammatory signal transduction pathways,
including the Janus kinase 2-signal transducer and activator of
transcription 3-suppressor of cytokine signaling 3 pathway
(14,36), the cyclic adenosine
3′,5′-monophosphate (cAMP) response element binding protein or the
cAMP-dependent protein kinase (19), which can inhibit the ASP-induced
activation and nuclear translocation of NF-κB.
The majority of reports state that p38 kinase can
positively regulate NF-κB activity, albeit through various
mechanisms (14,16,37).
By contrast, in certain studies, the inhibition of p38 kinase
significantly increased NF-κB activity (15,28).
Although examining the association between NF-κB and p38 kinase is
beyond the scope of the present study, it is important to determine
whether there is reciprocal cross-talk between NF-κB and p38 kinase
in the system, and further investigation of their interaction is
required. The present study may provide novel insights into obesity
treatment and various approaches toward the development of novel
anti-obesity therapeutic agents.
In conclusion, the results of the present study
suggested a novel anti-inflammatory function of α7nAChR in the
regulation of chemokine production by adipocytes in response to
ASP. Of note, α7nAChR appeared to exert its effects through
modulation of the p38 kinase pathway and the canonical NF-κB
pathway.
Acknowledgments
The authors would like to thank Mr Marc Lapointe
(Laval University, Québec, Canada) for the preparation and
purification of recombinant ASP. This study was supported by the
National Natural Science Foundation of China (grant no. 81300685)
to Dr Jing Wu (First Affiliated Hospital of Zhengzhou University,
Zhengzhou, China).
References
|
1
|
Coenen KR, Gruen ML, Chait A and Hasty AH:
Diet-induced increases in adiposity, but not plasma lipids, promote
macrophage infiltration into white adipose tissue. Diabetes.
56:564–573. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Neels JG, Badeanlou L, Hester KD and Samad
F: Keratinocyte-derived Chemokine in obesity: Expression,
regulation, and role in adipose macrophage infiltration and glucose
homeostasis. J Biol Chem. 284:20692–20698. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tom FQ, Gauvreau D, Lapointe M, Lu H,
Poursharifi P, Luo XP and Cianflone K: Differential chemoattractant
response in adipocytes and macrophages to the action of acylation
stimulating protein. Eur J Cell Biol. 92:61–69. 2013. View Article : Google Scholar
|
|
4
|
van Maanen MA, Papke RL, Koopman FA,
Koepke J, Bevaart L, Clark R, Lamppu D, Elbaum D, LaRosa GJ, Tak PP
and Vervoordeldonk MJ: Two novel α7 nicotinic acetylcholine
receptor ligands: In vitro properties and their efficacy in
collagen-induced arthritis in mice. PLoS One. 10:e01162272015.
View Article : Google Scholar
|
|
5
|
Peña G, Cai B, Liu J, van der Zanden EP,
Deitch EA, de Jonge WJ and Ulloa L: Unphosphorylated STAT3
modulates alpha7 nicotinic receptor signaling and cytokine
production in sepsis. Eur J Immunol. 40:2580–2589. 2010. View Article : Google Scholar
|
|
6
|
Kim TH, Kim SJ and Lee SM: Stimulation of
the α7 nicotinic acetylcholine receptor protects against sepsis by
inhibiting Toll-like receptor via phosphoinositide 3-kinase
activation. J Infect Dis. 209:1668–1677. 2014. View Article : Google Scholar
|
|
7
|
Marrero MB, Lucas R, Salet C, Hauser TA,
Mazurov A, Lippiello PM and Bencherif M: An alpha7 nicotinic
acetylcholine receptor-selective agonist reduces weight gain and
metabolic changes in a mouse model of diabetes. J Pharmacol Exp
Ther. 332:173–180. 2010. View Article : Google Scholar
|
|
8
|
Cancello R, Zulian A, Maestrini S,
Mencarelli M, Della Barba A, Invitti C, Liuzzi A and Di Blasio AM:
The nicotinic acetylcholine receptor α7 in subcutaneous mature
adipocytes: Downregulation in human obesity and modulation by
diet-induced weight loss. Int J Obes (Lond). 36:1552–1557. 2012.
View Article : Google Scholar
|
|
9
|
Berg AH, Lin Y, Lisanti MP and Scherer PE:
Adipocyte differentiation induces dynamic changes in NF-kappaB
expression and activity. Am J Physiol Endocrinol Metab.
287:E1178–E1188. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aggarwal BB: Targeting
inflammation-induced obesity and metabolic diseases by curcumin and
other nutraceuticals. Annu Rev Nutr. 30:173–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dandona P: Insulin resistance and
endothelial dysfunction in atherosclerosis: Implications and
interventions. Diabetes Technol Ther. 4:809–815. 2002. View Article : Google Scholar
|
|
12
|
He Z, Zhu HH, Bauler TJ, Wang J, Ciaraldi
T, Alderson N, Li S, Raquil MA, Ji K, Wang S, et al: Nonreceptor
tyrosine phosphatase Shp2 promotes adipogenesis through inhibition
of p38 MAP kinase. Proc Natl Acad Sci USA. 110:E79–E88. 2013.
View Article : Google Scholar :
|
|
13
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: A family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoon SW, Goh SH, Chun JS, Cho EW, Lee MK,
Kim KL, Kim JJ, Kim CJ and Poo H: alpha-Melanocyte-stimulating
hormone inhibits lipopolysaccharide-induced tumor necrosis
factor-alpha production in leukocytes by modulating protein kinase
A, p38 kinase and nuclear factor kappa B signaling pathways. J Biol
Chem. 278:32914–32920. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bowie AG and O'Neill LA: Vitamin C
inhibits NF-kappa B activation by TNF via the activation of p38
mitogen-activated protein kinase. J Immunol. 165:7180–7188. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feng M, Wang Y, Chen K, Bian Z, Jinfang Wu
and Gao Q: IL-17A promotes the migration and invasiveness of
cervical cancer cells by coordinately activating MMPs expression
via the p38/NF-κB signal pathway. PLoS One. 9:e1085022014.
View Article : Google Scholar
|
|
17
|
Saeed RW, Varma S, Peng-Nemeroff T, Sherry
B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y and Metz CN:
Cholinergic stimulation blocks endothelial cell activation and
leukocyte recruitment during inflammation. J Exp Med.
201:1113–1123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Jonge WJ and Ulloa L: The alpha7
nicotinic acetylcholine receptor as a pharmacological target for
inflammation. Br J Pharmacol. 151:915–929. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yoshikawa H, Kurokawa M, Ozaki N, Nara K,
Atou K, Takada E, Kamochi H and Suzuki N: Nicotine inhibits the
production of proinflammatory mediators in human monocytes by
suppression of I-kappaB phosphorylation and nuclear factor-kappaB
transcriptional activity through nicotinic acetylcholine receptor
alpha7. Clin Exp Immunol. 146:116–123. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun P, Zhou K, Wang S, Li P, Chen S, Lin
G, Zhao Y and Wang T: Involvement of MAPK/NF-κB signaling in the
activation of the cholinergic anti-inflammatory pathway in
experimental colitis by chronic vagus nerve stimulation. PLoS One.
8:e694242013. View Article : Google Scholar
|
|
21
|
Murray I, Parker RA, Kirchgessner TG, Tran
J, Zhang ZJ, Westerlund J and Cianflone K: Functional bioactive
recombinant acylation stimulating protein is distinct from C3a
anaphylatoxin. J Lipid Res. 38:2492–2501. 1997.
|
|
22
|
Shi D, Pop MS, Kulikov R, Love IM, Kung AL
and Grossman SR: CBP and p300 are cytoplasmic E4 polyubiquitin
ligases for p53. Proc Natl Acad Sci USA. 106:16275–16280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takahashi K, Yamaguchi S, Shimoyama T,
Seki H, Miyokawa K, Katsuta H, Tanaka T, Yoshimoto K, Ohno H,
Nagamatsu S and Ishida H: JNK- and IkappaB-dependent pathways
regulate MCP-1 but not adiponectin release from artificially
hypertrophied 3T3-L1 adipocytes preloaded with palmitate in vitro.
Am J Physiol Endocrinol Metab. 294:E898–E909. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martins RP, Kaur K, Hwang E, Ramirez RJ,
Willis BC, Filgueiras-Rama D, Ennis SR, Takemoto Y, Ponce-Balbuena
D, Zarzoso M, et al: Dominant frequency increase rate predicts
transition from paroxysmal to long-term persistent atrial
fibrillation. Circulation. 129:1472–1482. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang HF, Lin PP, Chen CH, Yeh YL, Huang
CC, Huang CY and Tsai CC: Effects of lactic acid bacteria on
cardiac apoptosis are mediated by activation of the
phosphatidylinositol-3 kinase/AKT survival-signalling pathway in
rats fed a high-fat diet. Int J Mol Med. 35:460–470. 2015.
|
|
26
|
Després JP: Body fat distribution and risk
of cardiovascular disease: An update. Circulation. 126:1301–1313.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fisette A, Poursharifi P, Oikonomopoulou
K, Munkonda MN, Lapointe M and Cianflone K: Paradoxical
glucose-sensitizing yet proinflammatory effects of acute ASP
administration in mice. Mediators Inflamm. 2013:7132842013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Weber NC, Blumenthal SB, Hartung T,
Vollmar AM and Kiemer AK: ANP inhibits TNF-alpha-induced
endothelial MCP-1 expression-involvement of p38 MAPK and MKP-1. J
Leukoc Biol. 74:932–941. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shytle RD, Mori T, Townsend K, Vendrame M,
Sun N, Zeng J, Ehrhart J, Silver AA, Sanberg PR and Tan J:
Cholinergic modulation of microglial activation by alpha 7
nicotinic receptors. J Neurochem. 89:337–343. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzuki T, Hide I, Matsubara A, Hama C,
Harada K, Miyano K, Andrä M, Matsubayashi H, Sakai N, Kohsaka S, et
al: Microglial alpha7 nicotinic acetylcholine receptors drive a
phospholipase C/IP3 pathway and modulate the cell activation toward
a neuroprotective role. J Neurosci Res. 83:1461–1470. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aicher A, Heeschen C, Mohaupt M, Cooke JP,
Zeiher AM and Dimmeler S: Nicotine strongly activates dendritic
cell-mediated adaptive immunity: Potential role for progression of
atherosclerotic lesions. Circulation. 107:604–611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang JZ, Liu Z, Liu J, Ren JX and Sun TS:
Mitochondrial DNA induces inflammation and increases TLR9/NF-κB
expression in lung tissue. Int J Mol Med. 33:817–824.
2014.PubMed/NCBI
|
|
33
|
Mishra NC, Rir-sima-ah J, Boyd RT, Singh
SP, Gundavarapu S, Langley RJ, Razani-Boroujerdi S and Sopori ML:
Nicotine inhibits Fc epsilon RI-induced cysteinyl leukotrienes and
cytokine production without affecting mast cell degranulation
through Alpha7/Alpha9/Alpha 10-nicotinic receptors. J Immunol.
2185:588–596. 2010. View Article : Google Scholar
|
|
34
|
Marrero MB and Bencherif M: Convergence of
alpha 7 nicotinic acetylcholine receptor-activated pathways for
anti-apoptosis and anti-inflammation: Central role for JAK2
activation of STAT3 and NF-kappaB. Brain Res. 1256:1–7. 2009.
View Article : Google Scholar
|
|
35
|
Chernyavsky AI, Arredondo J, Galitovskiy
V, Qian J and Grando SA: Upregulation of nuclear factor-kappaB
expression by SLURP-1 is mediated by alpha7-nicotinic acetylcholine
receptor and involves both ionic events and activation of protein
kinases. Am J Physiol Cell Physiol. 299:C903–C911. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kox M, van Velzen JF, Pompe JC,
Hoedemaekers CW, van der Hoeven JG and Pickkers P: GTS-21 inhibits
pro-inflammatory cytokine release independent of the Toll-like
receptor stimulated via a transcriptional mechanism involving JAK2
activation. Biochem Pharmacol. 78:863–872. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li X, Zhou X, Ye Y, Li Y, Li J, Privratsky
B, Wu E, Gao H, Huang C and Wu M: Lyn regulates inflammatory
responses in Klebsiella pneumoniae infection via the p38/NF-κB
pathway. Eur J Immunol. 44:763–773. 2014. View Article : Google Scholar
|