Introduction
Optimal oxygen supply is of the utmost importance
for the effective functioning of cells with high energy demand,
including cardiomyocytes and myocytes, since their metabolism
relies on the oxidative pathway (1–3).
Nonetheless, their productive performance is critically contributed
to by another agent, namely iron (4). As an essential component of
co-factors present both in oxygen carriers and in numerous enzymes
of oxidative metabolism, iron constitutes the cornerstone of tissue
oxygen supply and energy generation (4–7).
Notably, the experimental data demonstrates that hypoxia and an
iron deficient state may have to some extent, similar consequences
in both cardiac and skeletal myocytes at the molecular level,
including impairment of the mitochondrial electron transport chain
and triggering a shift from oxidative to glycolytic metabolism
(8–13).
Previous evidence suggests that the optimal iron
homeostasis is crucial for patients with various cardiovascular
disorders, including coronary artery disease (14) and heart failure (15–17),
in which hypoxia is an important causative factor. Notably, data
from previous clinical studies confirmed the beneficial effects of
iron therapy in non-anaemic iron-deficient patients with heart
failure accompanied by skeletal myopathy (18–23).
To date, the majority of studies have investigated the molecular
links between hypoxia and iron metabolism in different cells
involved in systemic iron homeostasis, including macrophages,
enterocytes, hepatocytes or erythroid precursor cells (24–28);
however, very little attention have been given to gain an insight
into molecular associations between oxygen and iron availability in
the cells of cardiac or skeletal muscle tissues. Robach et
al (29) investigated the
influence of hypoxia on the expression of selected genes of iron
metabolism, namely transferrin receptor and L-ferritin (FTH), in
skeletal muscle; however, the studies involved human subjects
exposed to high-altitude hypoxia where it was difficult to obtain
changing experimental conditions.
Since hypoxic conditions are difficult to introduce
and manipulate in humans and animals, the present study used cell
culture models. The aim of the present study was to investigate the
associations between selected genes involved in iron metabolism and
cellular apoptotic activity assessed in diverse oxygen and iron
availabilities in two rat cell lines: Cardiomyocytes (H9C2) and
skeletal myocytes (L6G8C5).
Materials and methods
Cell culture
Rat H9C2 cardiomyocytes and rat L6G9C2 (L6) skeletal
myocytes (Sigma-Aldrich, St. Louis, MO, USA) were grown in
Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich),
supplemented with 10% fetal bovine serum (FBS; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 2 mmol/l glutamine,
104 diluted 10,000 U/ml penicillin and 10 mg/ml
streptomycin (all from Sigma-Aldrich). For passaging, the cells
were washed with phosphate-buffered saline (PBS; without
Ca2+ and Mg2+) and were released by
trypsinization (Sigma-Aldrich). The cells were maintained according
to manufacturer's protocols.
Experimental schedule
Rat H9C2 cardiomyocytes and rat L6G9C2 skeletal
myocytes were cultured with an addition of deferoxamine (DFO;
Sigma-Aldrich) or ammonium ferric citrate (AFC; Sigma-Aldrich) in
order to change iron accessibility in hypoxia for those cells in
the cultured medium during hypoxia treatment for 48 h. DFO is a
selective iron chelator commonly used in the cell culture studies.
It has been reported that addition of DFO into the culture medium
reduces iron concentration both in the cellular environment and
inside the cell, since DFO can be taken up by fluid phase
endocytosis (30). AFC was applied
in cell culture studies in order to induce intracellular iron
accumulation (31,32).
Modulation of iron concentration in the
cellular environment
H9C2 and L6 cells were seeded at 0.3×106
cells/well in a volume of 2 ml in 6-well plates. The cells were
cultured under hypoxic conditions (1% O2, 5%
CO2, 94% N2), which were generated in a
standard cell culture incubator by displacing O2 with
infusion of N2, which was supplied by an external
high-pressure liquid nitrogen tank, in differential iron
availability in the growth media for 48 h. The cells were separated
into three groups: i) Optimal iron concentration (standard iron
concentration in DMEM with 10% FBS); ii) reduced iron concentration
(iron chelation using 100 µM DFO); iii) increased iron
concentration (supplementation with 200 µM AFC). The compounds were
added to the cells from 1,000X concentrated stocks diluted in
culture medium.
Assessed parameters
Multiple parameters were assessed during the present
study. These parameters reflected the cellular condition and iron
status assessed in the states of different iron availability in
culture medium of H9C2 and L6 lines. These parameters were looking
at the expression levels of various genes in the following
processes: i) Apoptosis, including B-cell lymphoma
(Bcl)-2-associated X protein (Bax; inductor) (33), Bcl-2 (inhibitor)
(34) and the Bax/Bcl-2 gene
expression ratio (35); ii)
proteolysis, including Atrogin-1, which is a marker of protein
degradation in myocytes and a marker of muscle atrophy (36); iii) glycolysis, including pyruvate
kinase (PKM2; marker of non-oxidative metabolism) (37); iv) intracellular iron metabolism,
including transferrin receptor type 1 (TfR1; cellular iron
importer) (38), ferroportin
(FPN1; cellular iron exporter) (39), FTH heavy chain (iron storage
protein) (40) and hepcidin
(HAMP; iron metabolism regulator protein) (41).
Cell viability tetrazolium reduction
assay (MTS)
The MTS assay is a colorimetric method used to
determine the number of viable and metabolically active cells in
proliferation and cytotoxicity assays, as previously described
(42). MTS assays were performed,
according to manufacturer's protocol (CellTiter 96®
AQueous One Solution Cell Proliferation Assay; Promega Corporation,
Madison, WI, USA). Briefly, 2×105 H9C2 or L6 cells were
seeded into each well of 96-well plates and were treated for 48 h
with DFO, AFC or PBS (control) in hypoxia or in normoxia, as
described above. A total of 20 µl CellTiter 96® AQueous
One Solution reagent was added to each well and the absorbance at
490 nm was measured after 2 h incubation in 37°C (KCjunior™; BioTek
Instruments, Inc., Winooski, VT, USA). The viability of the control
cells was treated as 100%.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The total RNA was prepared from H9C2 or L6 cells
harvested from 6-well tissue culture plates using the RNeasy
Fibrous Tissue Mini kit (Qiagen, Hilden, Germany), according to the
manufacturer's protocol. The protocol included an on-column DNase
digestion to remove the genomic DNA. First-strand cDNA was
synthesized using a SuperScript III First-Strand Synthesis system
with oligo(dT)20 primer (Invitrogen; Thermo Fisher Scientific,
Inc.).
Based on the rat genomic and cDNA sequences the
following primers were used: Bcl-2, forward:
5′-AGCATGCGACCTCTGTTTGA-3′ and reverse 5′-TCACTTGTGGCCCAGGTATG-3′;
Bax, forward: 5′-TGGCGATGAACTGGACAACA-3′ and reverse:
5′-CACGGAAGAAGACCTCTCGG-3′; Atrogin-1 forward:
5′AGCTTGTGCGATGTTACCCA-3′ and reverse 5′-GAGCAGCTCTCTGGGTTGTT-3′;
PKM2, forward: 5′-TTAGGCCAGCAACGCTTGTA-3′ and reverse:
5′-AGCTGGGCTCTATTGCATGT-3′; TfR1, forward:
5′-GAGACTACTTCCGTGCTACTTC-3′ and reverse:
5′-TGGAGATACATAGGGTGACAGG-3′; FPN1, forward:
5′-TCGGTCTTTGGTCCTTTGATTTG-3′ and reverse
5′-GGCTGACTTTCATCTGTAACTTCC-3′; FTH, forward:
5′-GCCAAATACTTTCTCCATCAATCTC-3′ and reverse:
5′-CCGCTCTCCCAGTCATCAC-3′; HAMP, forward:
5′-GCAACAGACGAGACAGACTAC-3′ and reverse 5′-GCAACAGAGACCACAGGAG-3′.
The primers were designed with Molecular Beacon Software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The primers spanned exon
junctions to prevent the amplification of genomic DNA. The
reference genes, Actb and 18S-RNA, were
selected for normalization of experimental conditions by means of
geNorm analysis (PrimerDesign Ltd., Southampton, UK).
All samples were performed in triplicates. The qPCR
assay was performed on the CFX Connect Real-Time PCR Detection
system. The specificity of PCR was determined by melting curve
analysis for each reaction. The amplification efficiency was
estimated by running serial dilutions of a template. Successive
dilutions were plotted against the appropriate Cq values to
generate a standard curve. The slope calculated from the standard
curve was used to determine the amplification efficiency (E)
according to the formula: E=10-1/slope. The amplification
efficiencies for the target amplicons, actb and
18srna, were not comparable and the Pfaffl method was used
to determine the relative expression (43).
Statistical analysis
The data are presented as the mean ± standard
deviation, unless otherwise indicated. Mann-Whitney U Test
was used to compare the groups. All molecular assessments were
performed in triplicates. R-value is a correlation coefficient
between the expression of iron metabolism genes in three states of
iron concentrations and the Bax/Bcl-2 gene
expression ratio in those conditions. P<0.05 was considered to
indicate a statistically significant difference.
Results
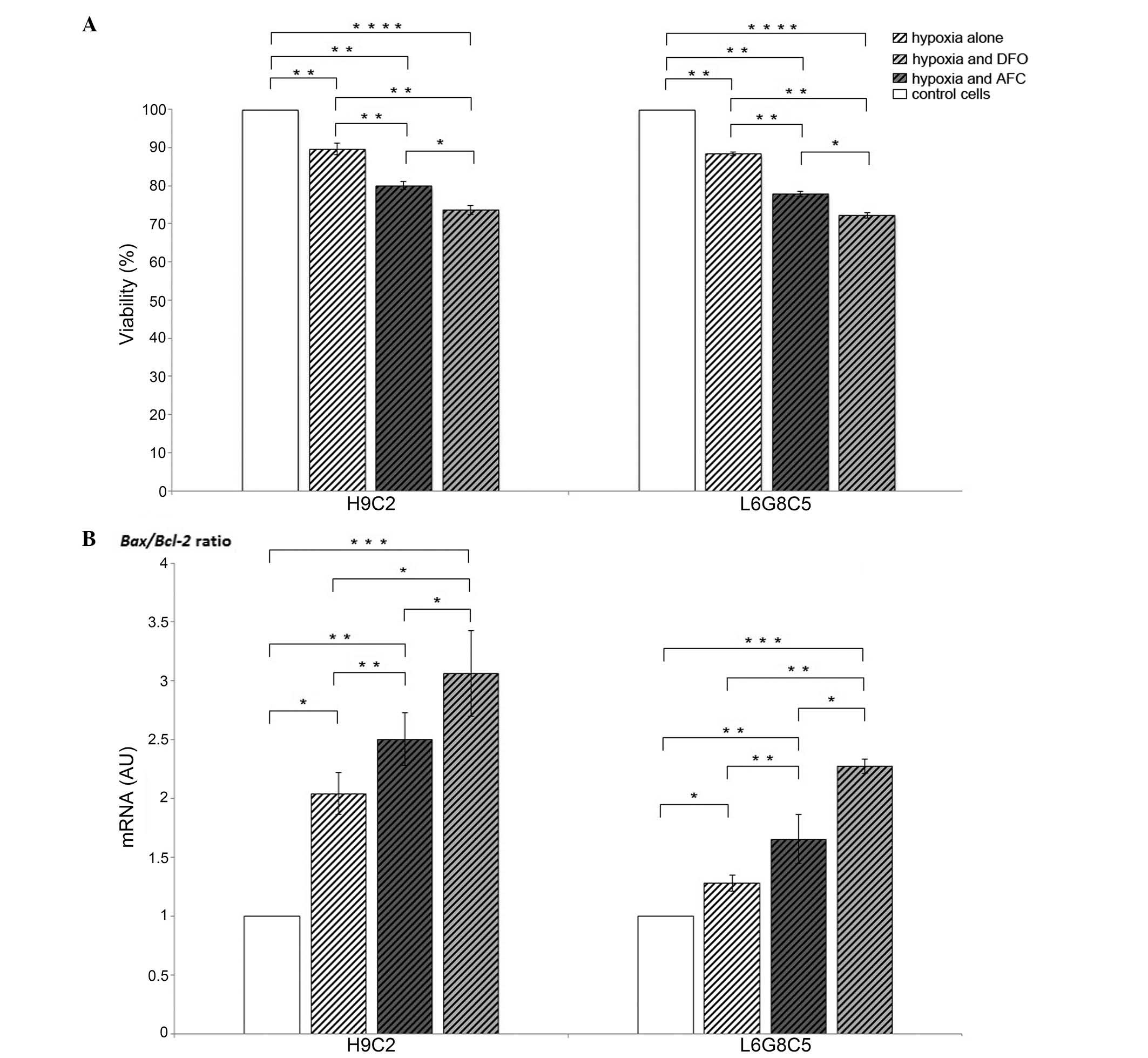
Effects of hypoxia on the viability and
apoptotic activity of cardiomyocytes and myocytes
Low oxygen treatment reduced the viability of
cardiomyocytes and myocytes by 11 and 12%, respectively, as
compared with the untreated cells (P<0.01 each). In each cell
type, the decrement in viability was associated with an increased
Bax/Bcl-2 gene expression ratio (P<0.05),
reflecting an enhanced susceptibility of cardiomyocytes and
myocytes to cellular programmed death (Fig. 1).
Bax/Bcl-2 ratio is associated with the
loss of viability of cardiomyocytes and myocytes during hypoxia
with concomitant reduced or increased iron availability
In both cardiomyocytes and myocytes, the exposition
to low iron availability during hypoxia caused, as compared with
the cells cultured during hypoxia and optimal iron concentration, a
15% loss in viability in each of the cell lines (P<0.01;
Fig. 1A), and this change was
associated with an increase in Bax/Bcl-2 gene
expression ratio (P<0.001; Fig.
1B). Likewise, both H9C2 and L6 cells demonstrated an increase
in Bax/Bcl-2 gene expression ratio (P<0.05
and P<0.01; Fig. 1B) during
hypoxia, when exposed to the elevated iron availability, which was
associated with 10 and 11% loss in viability in cardiomyocytes and
myocytes, respectively (P<0.01; Fig. 1A). Notably, in each cell type
during hypoxia, the increase in the Bax/Bcl-2
ratio (Fig. 1B) and associated
viability loss (Fig. 1A) were
greater upon exposure to low iron availability compared with that
in the case of AFC treatment (P<0.05 each).
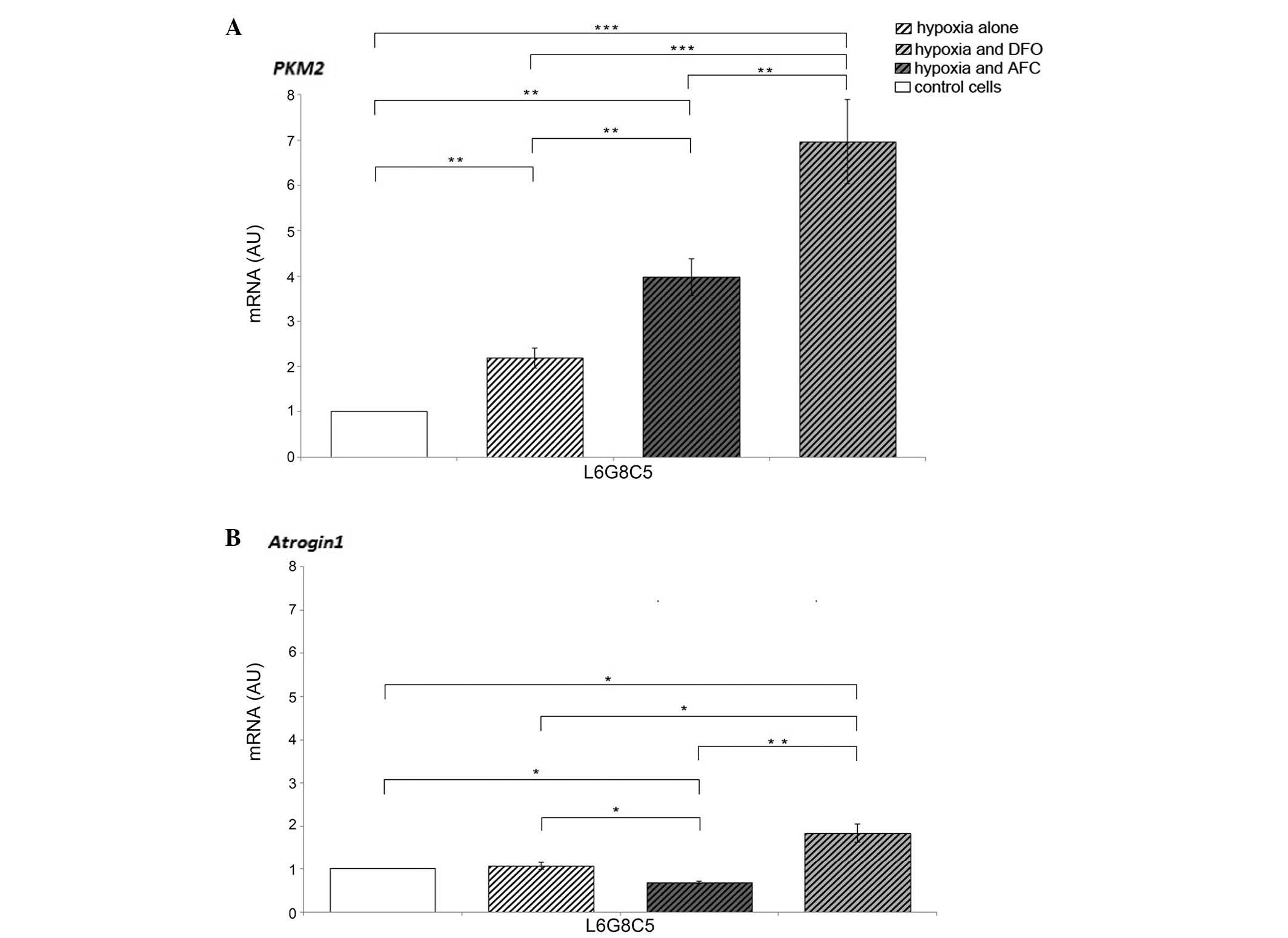
Effects of hypoxia with concomitant
optimal, reduced or increased iron availability on tissue-specific
expression of genes involved in energy metabolism and proteolysis
in myocytes
Hypoxic conditions of L6 cell culture caused an
increase in the expression of PKM2, which responsible for
the rate-limiting step of glycolysis, when compared with cells
cultured in normoxia (P<0.01). Myocytes exposed to DFO during
hypoxia demonstrated increased mRNA expression levels of
PKM2 and Atrogin-1 (P<0.001 and P<0.05,
respectively) as compared with the cells cultured in normal iron
concentration, suggesting the further enhancement of non-oxidative
glycolytic pathway and protein degradation in the cells. The
different changes were observed in myocytes treated with AFC, since
the increase in the expression of PKM2 (P<0.01) was
accompanied by a decrease in the mRNA expression of
Atrogin-1 (P<0.05) as compared with the cells
cultured in optimal iron concentration. The augmented expression of
PKM2 in myocytes during hypoxia was greater upon low iron
availability compared with during ferric salt treatment
(P<0.01). Notably, the expression of Atrogin-1
during hypoxia and concomitant elevated iron availability was
decreased as compared with cells cultured in hypoxia and normal or
decreased iron concentration (P<0.05 and P<0.01,
respectively; Fig. 2).
Effects of hypoxia with concomitant
optimal, reduced or increased iron availability on the expression
of genes involved in iron influx and efflux in cardiomyocytes and
myocytes
Both L6 and H9C2 cell lines exposed to hypoxia
demonstrated, as compared with the cells cultured in normoxia, a
decrease in the expression TfR1 (P<0.05 each) and
FPN1 (P<0.05 and P<0.01, respectively).
The addition of DFO to the culture medium of
cardiomyocytes and myocytes during hypoxia caused an increased
expression of TfR1 (P<0.01 and P<0.001, respectively),
reflecting iron demand, thus facilitated entrance of iron to the
cells. However, this did not affect the expression of FPN1
compared with the cells cultured in normal iron concentration.
The AFC treatment of cardiomyocytes and myocytes
during hypoxia resulted in a decreased and increased, respectively,
expression of TfR1 (P<0.05 each). Additionally, this
resulted in an increased expression of FPN1 (P<0.05 each)
in each cell line. Under hypoxic conditions with concomitant
changing iron availability, the myocytes demonstrated low
expression of FPN1 when compared with the cells cultured in
normoxia (P<0.01) (Fig. 3A and
B).
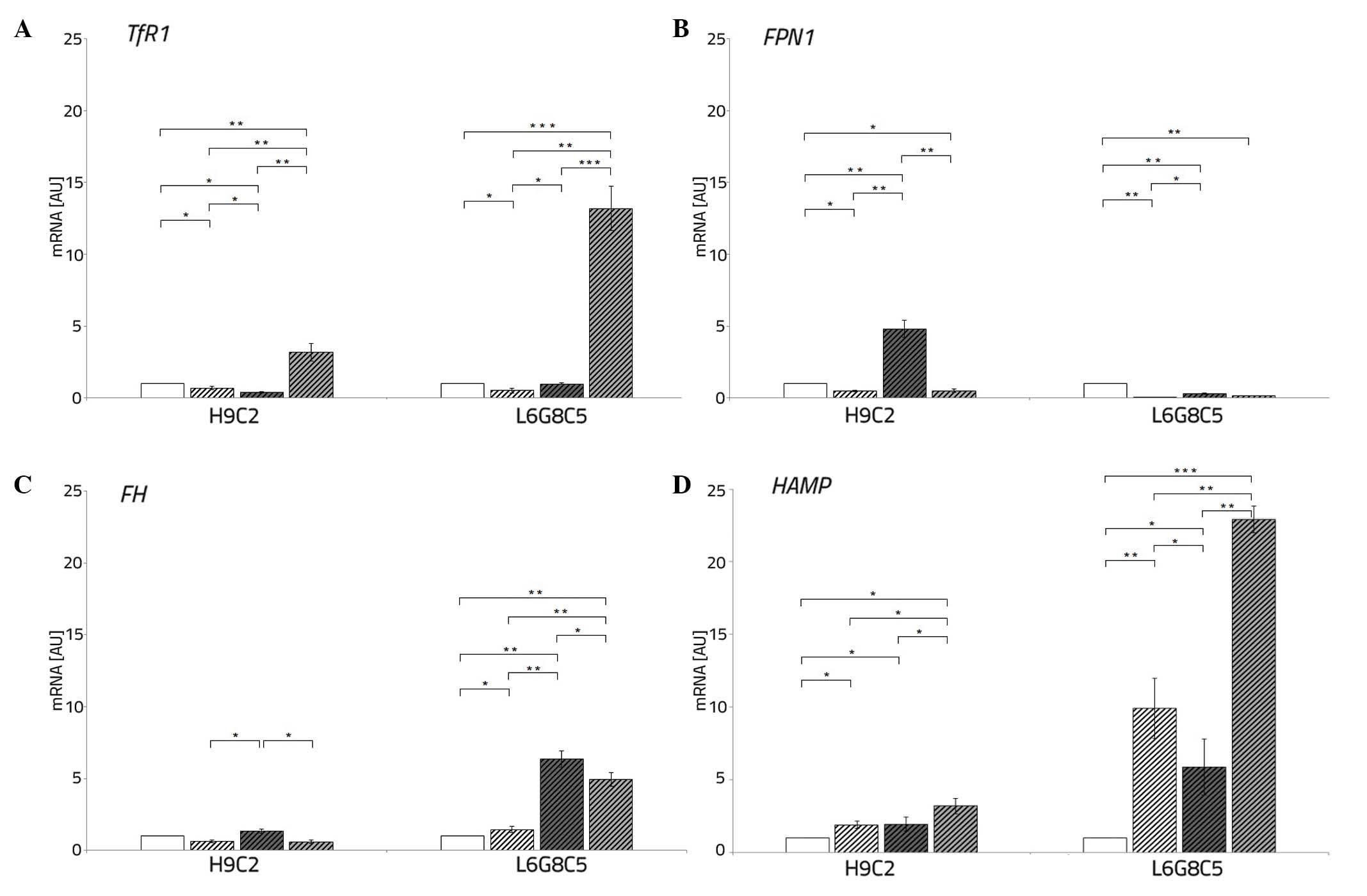 | Figure 3Expression of iron metabolism genes
in L6G8C5 and H9C2 cells exposed to hypoxia alone, hypoxia and DFO
or hypoxia and AFC for 48 h. The expression levels of (A)
TfR1, (B) FPN1, (C) FTH and (D) HAMP
were assessed. Control cells were cultured in normoxia and were not
treated with AFC or DFO. The data are presented as the
mean±standard deviation (*P<0.05;
**P<0.01; ***P<0.001, compared with the
control). DFO, deferoxamine; AFC, ammonium ferric citrate; TfR1,
transferrin receptor type 1; FPN1, ferroportin type 1; FTH,
ferritin heavy chain; HAMP, hepcidin. |
Effects of hypoxia with concomitant
optimal, reduced or increased iron availability on the expression
of the iron storage gene in cardiomyocytes and myocytes
Low oxygen treatment of L6 cells caused, as compared
with cells cultured in normoxia, an increased expression of the
iron storage gene, FTH (P<0.05). Exposure to DFO of
cardiomyocytes during hypoxia resulted, as compared with
hypoxia-treated controls, in unchanged mRNA expression of
FTH, while hypoxic culture of L6 cells exposed to low iron
demonstrated an increase in the expression of the iron storage gene
(P<0.01).
The different pattern of changes was observed upon
ferric salt treatment during hypoxia. An increased expression of
FTH in both cardiomyocytes and myocytes was observed
compared with the cells cultured in normal iron concentration
(P<0.05 and P<0.01, respectively) (Fig. 3C).
Effects of hypoxia with concomitant
optimal, reduced or increased iron availability on the expression
of iron regulator in cardiomyocytes and myocytes
Low oxygen treatment of H9C2 and L6 cells caused an
increment in the expression of iron regulator, HAMP
(P<0.05 and P<0.01, respectively). An addition of DFO to the
culture medium of both studied cell lines during hypoxia caused a
further increase in the expression of HAMP (P<0.05 and
P<0.001) as compared with the cells cultured in a normal iron
concentration. The exposition of cardiomyocytes and myocytes to AFC
treatment in hypoxia resulted in an unchanged or a decreased
expression of HAMP (P<0.01) (Fig. 3D).
An increased expression of iron-related
genes in a low iron state during hypoxia is associated with an
increased apoptotic activity of cardiomyocytes and myocytes
In both cardiomyocytes and myocytes, the expression
of genes involved in iron influx (TfR1) and regulation
(HAMP) was associated with the
Bax/Bcl-2 gene expression ratio in the whole
spectrum of iron status during hypoxia (all R>0.6, P<0.05).
In particular, in a low iron state, the observed upregulation of
TfR1 and HAMP genes was associated with increased
apoptotic activity of each of the cell lines. In addition, L6 cells
demonstrated positive correlation between the expression of
FTH and the Bax/Bcl-2 gene expression
ratio (R=0.64, P<0.05) (Fig.
4).
Discussion
The present study compared the influence of
increased or reduced iron availability during hypoxia on the
viability and apoptotic activity of the cardiac and skeletal
myocytes. It was discovered that during hypoxia, the reduced iron
concentration had a more negative impact on the viability and
functioning of the studied myocytes compared with the elevated iron
availability. Furthermore, in these cells cultured during hypoxia
and with differential iron availability, the present study was able
to investigate changes in the expression of iron turnover genes and
to establish associations between the upregulated expression of
genes involved in iron influx (TfR1) and regulation
(HAMP), and the enhanced susceptibility to cellular
suicide.
Firstly, the present study demonstrated that hypoxic
conditions diminished the viability of cardiomyocytes and myocytes.
The accompanying changes in the expression of genes involved in the
regulation of apoptosis supported the results from the viability
assay, since the gene expression ratio of pro-apoptotic Bax
to anti-apoptotic Bcl-2 increased upon low oxygen.
Furthermore, it was demonstrated that the exposition of the studied
cells to either iron depletion or iron excess during hypoxia
hampered the cellular viability and increased
Bax/Bcl-2 gene expression ratio more severely
compared with hypoxia treatment alone. This finding was consistent
with the study of Walter et al (44) who demonstrated that both iron
overload and iron deficiency are detrimental to the functioning of
liver mitochondria (44). With
this work, besides highlighting the presence of analogous U-shaped
association between iron availability and vitality of the studied
cells during hypoxia, the present study identified that upon low
oxygen conditions, iron deficiency has a more negative impact on
the cellular viability and with a greater extent favors apoptosis
rather than iron excess.
To the best of our knowledge, the local iron
metabolism has only been partially assessed in the cell culture
models of myocardium and skeletal muscles (43,44).
It remains insufficiently investigated upon hypoxia conditions; to
date it was explored in skeletal muscle on the model of tissue
samples from patients subjected to high-altitude hypoxia (29). The present results are consistent
with the study of Robach et al (29), as we observed the hypoxia-driven
decrease in the mRNA expression of TfR1 and increase in the
mRNA expression of FTH. However, the present model of cell
cultures allowed manipulations within the oxygen and iron
availabilities, which were not amenable in humans. Therefore, the
present study has provided the first evidence, to the best of our
knowledge, of the direct response of iron turnover genes to hypoxia
and changing iron availability within the isolated model of
cardiomyocytes and skeletal myocytes cultured in vitro.
It was shown that the exposure of cardiomyocytes and
myocytes to low oxygen altered the expression of iron turnover
genes in each of the cell lines. Notably, these changes vary from
the previously reported data, since to date, the studies on the
expression of iron-related genes during hypoxia have been performed
predominantly in the cells involved in systemic iron homeostasis,
including hepatocytes, macrophages, enterocytes or erythroid
precursor cells (24–28). For example, the responses of the
aforementioned cells to low oxygen involved a hypoxia-driven
upregulation of both TfR1, in order to enable an enhanced
iron uptake by erythrocytes, and FPN1, resulting in the
enhanced release of iron into the bloodstream from iron storing and
distributing cells (47–49). Furthermore, hypoxia was
demonstrated to downregulate the expression of HAMP and to
increase the expression of FTH in hepatocytes (50–52).
As mentioned previously, the expression changes that
occurred during hypoxia within cardiomyocytes and skeletal
myocytes, which both represent tissues of minor importance in
respect to systemic iron homeostasis, are very often inconsistent
with data previously obtained from the other cell types. In hypoxic
conditions, both studied cell lines have demonstrated a decrease in
the expression of transferrin receptor and this change was
accompanied with a decreased expression of ferroportin gene.
Notably, unlike the previous data reporting hypoxia-driven
decrement of hepatic hepcidin expression, the present finding
demonstrated in both cardiomyocytes and myocytes during hypoxia a
markedly increased expression of hepcidin. It is worthwhile
mentioning that the function of this iron regulatory hormone
locally expressed in the heart and skeletal muscle remains to be
fully understood (53–55) and in depth research in this field
is required. Furthermore, in skeletal myocytes, the alteration of
the expression of iron storage protein, FTH, which was
increased in low oxygen was observed. This change may be associated
with the potential antioxidant role of FTH in the protective
mechanism of myocytes against the hypoxia-mediated generation of
reactive oxygen species (51,52,56).
In addition, the present study has demonstrated that
the treatment with an iron chelator upon hypoxia resulted in an
increased expression of transferrin receptor gene in both
cardiomyocytes and myocytes. This finding indicated that the
environmental iron depletion, along with low oxygen, may induce an
adaptive reaction of the cells, which enables iron resorption.
Notably, increased expression of TfR was also demonstrated,
reflecting a cellular iron starvation, suggesting an association
with the elevated Bax/Bcl-2 gene expression
ratio, thus increased apoptosis. Both cell lines, when exposed to
DFO in hypoxia, demonstrated a markedly increased level of
HAMP and this change was associated with the elevated
Bax/Bcl-2 gene expression ratio. Therefore, it
may be hypothesized that locally expressed hepcidin may account for
an important agent in stress conditions, including hypoxia and/or
iron deficiency.
In the case of skeletal myocytes, several issues are
worthy of consideration. Firstly, hypoxia and concomitant either
reduced or increase iron availability caused an increased
expression of the PKM2 gene, thus, the enhancement of
anaerobic glycolysis. This finding suggested that myocytes
underwent an oxidative-to-glycolytic shift in the experimental
conditions. These observations are consistent with the previously
reported data on the influence of hypoxia or iron deficiency on
skeletal muscle metabolism (8,9).
However, the present study has confirmed that the elevation of iron
concentration during hypoxia favors glycolysis to a lesser extent
compared with iron limitation. Furthermore, it was demonstrated
that in hypoxic conditions the increased iron concentration may be
to a certain extent protective since the expression of the marker
of muscle atrophy, Atrogin-1, was decreased upon
elevated iron availability in comparison with hypoxia alone or
hypoxia and iron chelator treatment. Notably,
Atrogin-1 expression indicated the imbalance between
anabolic and catabolic processes in the myocytes and favored
protein breakdown. It appears that during hypoxia the elevated iron
availability may suppress muscle atrophy, which results in the loss
of muscle mass and leads to muscle weakness, inactivity and
increased mortality (36).
Taking together, the present preliminary data
suggested that during hypoxia, iron excess is less detrimental for
the vitality of cardiomyocytes and skeletal myocytes, compared with
in an iron deficiency situation. In addition, it may be
hypothesized that the increased availability of iron during hypoxia
may be beneficial to a certain extent for skeletal myocytes in the
context of preventing muscle atrophy. This premise may stand for a
molecular substantiation of the efficacy of iron therapy for the
improvement of muscle functional capacity (e.g. in patients with
heart failure and concomitant iron deficiency).
Notably, it may be interesting to further
investigate the role of the locally expressed hepcidin in
cardiomyocytes and skeletal myocytes since the pattern of its
expression differs from what has already been explored in
hepatocytes.
Abbreviations:
|
PKM2
|
pyruvate kinase isoform 2
|
|
TfR1
|
transferrin receptor type 1
|
|
FPN1
|
ferroportin type 1
|
|
HAMP
|
hepcidin
|
|
FTH
|
ferritin heavy chain
|
|
DFO
|
deferoxamine
|
|
AFC
|
ammonium ferric citrate
|
Acknowledgments
The present study was supported by a research grant
from the Polish Ministry of Science and Higher Education (no.
UMO-2012/05/E/NZ5/00590).
References
|
1
|
Ferrari M, Binzoni T and Quaresima V:
Oxidative metabolism in muscle. Philos Trans R Soc Lond B Biol Sci.
352:677–683. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Atalay M and Hänninen OOP: Muscle energy
metabolism. Atalay M and Hänninen O: Encyclopedia of Life Support
Systems: Physiology and Maintenance. IV. Eolss Publishers Company
Limited; pp. 26–47. 2009
|
|
3
|
Stanley WC, Recchia FA and Lopaschuk GD:
Myocardial substrate metabolism in the normal and failing heart.
Physiol Rev. 85:1093–1129. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hower V, Mendes P, Torti FM, Laubenbacher
R, Akman S, Shulaev V and Torti SV: A general map of iron
metabolism and tissue-specific subnetworks. Mol Biosyst. 5:422–443.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Andrews NC: Disorders of iron metabolism.
N Engl J Med. 341:1986–1995. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Beard JL: Iron biology in immune function,
muscle metabolism and neuronal functioning. J Nutr. 131:568S–579S;
discussion 580S. 2001.PubMed/NCBI
|
|
7
|
Galy B, Ferring-Appel D, Sauer SW, Kaden
S, Lyoumi S, Puy H, Kölker S, Gröne HJ and Hentze MW: Iron
regulatory proteins secure mitochondrial iron sufficiency and
function. Cell Metab. 12:194–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ambrose LJ, Abd-Jamil AH, Gomes RS, Carter
EE, Carr CA, Clarke K and Heather LC: Investigating mitochondrial
metabolism in contracting HL-1 cardiomyocytes following hypoxia and
pharmacological HIF activation identifies HIF-dependent and
independent mechanisms of regulation. J Cardiovasc Pharmacol Ther.
19:574–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ohlendieck K: Proteomic identification of
biomarkers of skeletal muscle disorders. Biomark Med. 7:169–186.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Finch CA, Miller LR, Inamdar AR, Person R,
Seiler K and Mackler B: Iron deficiency in the rat. Physiological
and biochemical studies of muscle dysfunction. J Clin Invest.
58:447–453. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Davies KJ, Maguire JJ, Brooks GA, Dallman
PR and Packer L: Muscle mitochondrial bioenergetics, oxygen supply,
and work capacity during dietary iron deficiency and repletion. Am
J Physiol. 242:E418–E427. 1982.PubMed/NCBI
|
|
12
|
Henderson SA, Dallman PR and Brooks GA:
Glucose turnover and oxidation are increased in the iron-deficient
anemic rat. Am J Physiol. 250:E414–E421. 1986.PubMed/NCBI
|
|
13
|
Thompson CH, Green YS, Ledingham JG, Radda
GK and Rajagopalan B: The effect of iron deficiency on skeletal
muscle metabolism of the rat. Acta Physiol Scand. 147:85–90. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ponikowska B, Suchocki T, Paleczny B,
Olesinska M, Powierza S, Borodulin-Nadzieja L, Reczuch K, von
Haehling S, Doehner W, Anker SD, et al: Iron status and survival in
diabetic patients with coronary artery disease. Diabetes Care.
36:4147–4156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okonko DO, Mandal AK, Missouris CG and
Poole-Wilson PA: Disordered iron homeostasis in chronic heart
failure: Prevalence, predictors, and relation to anemia, exercise
capacity, and survival. J Am Coll Cardiol. 58:1241–1251. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jankowska EA, Rozentryt P, Witkowska A,
Nowak J, Hartmann O, Ponikowska B, Borodulin-Nadzieja L, Banasiak
W, Polonski L, Filippatos G, et al: Iron deficiency: An ominous
sign in patients with systolic chronic heart failure. Eur Heart J.
31:1872–1880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jankowska EA, Malyszko J, Ardehali H,
Koc-Zorawska E, Banasiak W, von Haehling S, Macdougall IC, Weiss G,
McMurray JJ, Anker SD, et al: Iron status in patients with chronic
heart failure. Eur Heart J. 34:827–834. 2013. View Article : Google Scholar :
|
|
18
|
Bolger AP, Bartlett FR, Penston HS,
O'Leary J, Pollock N, Kaprielian R and Chapman CM: Intravenous iron
alone for the treatment of anemia in patients with chronic heart
failure. J Am Coll Cardiol. 48:1225–1227. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Toblli JE, Lombraña A, Duarte P and Di
Gennaro F: Intravenous iron reduces NT-pro-brain natriuretic
peptide in anemic patients with chronic heart failure and renal
insufficiency. J Am Coll Cardiol. 50:1657–1665. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Anker SD, Comin Colet J, Filippatos G,
Willenheimer R, Dickstein K, Drexler H, Lüscher TF, Bart B,
Banasiak W, Niegowska J, et al: Ferric carboxymaltose in patients
with heart failure and iron deficiency. N Engl J Med.
361:2436–2448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ponikowski P, van Veldhuisen DJ,
Comin-Colet J, Ertl G, Komajda M, Mareev V, McDonagh T, Parkhomenko
A, Tavazzi L, Levesque V, et al: Beneficial effects of long-term
intravenous iron therapy with ferric carboxymaltose in patients
with symptomatic heart failure and iron deficiency†. Eur Heart J.
36:657–668. 2015. View Article : Google Scholar
|
|
22
|
Okonko DO, Grzeslo A, Witkowski T, Mandal
AK, Slater RM, Roughton M, Foldes G, Thum T, Majda J, Banasiak W,
et al: Effect of intravenous iron sucrose on exercise tolerance in
anemic and nonanemic patients with symptomatic chronic heart
failure and iron deficiency FERRIC-HF: A randomized, controlled,
observer-blinded trial. J Am Coll Cardiol. 51:103–112. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Beck-da-Silva L, Piardi D, Soder S, Rohde
LE, Pereira-Barretto AC, de Albuquerque D, Bocchi E, Vilas-Boas F,
Moura LZ, Montera MW, et al: IRON-HF study: A randomized trial to
assess the effects of iron in heart failure patients with anemia.
Int J Cardiol. 168:3439–3442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramey G, Deschemin JC, Durel B,
Canonne-Hergaux F, Nicolas G and Vaulont S: Hepcidin targets
ferroportin for degradation in hepatocytes. Haematologica.
95:501–504. 2010. View Article : Google Scholar :
|
|
25
|
Kuriyama-Matsumura K, Sato H, Yamaguchi M
and Bannai S: Regulation of ferritin synthesis and iron regulatory
protein 1 by oxygen in mouse peritoneal macrophages. Biochem
Biophys Res Commun. 249:241–246. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lok CN and Ponka P: Identification of a
hypoxia response element in the transferrin receptor gene. J Biol
Chem. 274:24147–24152. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cianetti L, Segnalini P, Calzolari A,
Morsilli O, Felicetti F, Ramoni C, Gabbianelli M, Testa U and Sposi
NM: Expression of alternative transcripts of ferroportin-1 during
human erythroid differentiation. Haematologica. 90:1595–1606.
2005.PubMed/NCBI
|
|
28
|
Peyssonnaux C, Nizet V and Johnson RS:
Role of the hypoxia inducible factors HIF in iron metabolism. Cell
Cycle. 7:28–32. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robach P, Cairo G, Gelfi C, Bernuzzi F,
Pilegaard H, Viganò A, Santambrogio P, Cerretelli P, Calbet JA,
Moutereau S and Lundby C: Strong iron demand during hypoxia-induced
erythropoiesis is associated with down-regulation of iron-related
proteins and myoglobin in human skeletal muscle. Blood.
109:4724–4731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Woo KJ, Lee TJ, Park JW and Kwon TK:
Desferrioxamine, an iron chelator, enhances HIF-1alpha accumulation
via cyclooxy-genase-2 signaling pathway. Biochem Biophys Res
Commun. 343:8–14. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parkes JG, Hussain RA, Olivieri NF and
Templeton DM: Effects of iron loading on uptake, speciation, and
chelation of iron in cultured myocardial cells. J Lab Clin Med.
122:36–47. 1993.PubMed/NCBI
|
|
32
|
Hoepken HH1, Korten T, Robinson SR and
Dringen R: Iron accumulation, iron-mediated toxicity and altered
levels of ferritin and transferrin receptor in cultured astrocytes
during incubation with ferric ammonium citrate. J Neurochem.
88:1194–1202. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakuragi N, Salah-eldin AE, Watari H, Itoh
T, Inoue S, Moriuchi T and Fujimoto S: Bax, Bcl-2, and p53
expression in endometrial cancer. Gynecol Oncol. 86:288–96. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–74. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Aliparasti MR, Alipour MR, Almasi S and
Feizi H: Ghrelin administration increases the Bax/Bcl-2 gene
expression ratio in the heart of chronic hypoxic rats. Adv Pharm
Bull. 5:195–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bodine SC and Baehr LM: Skeletal muscle
atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am
J Physiol Endocrinol Metab. 307:E469–E484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takenaka M, Noguchi T, Inoue H, Yamada K,
Matsuda T and Tanaka T: Rat pyruvate kinase M gene. Its complete
structure and characterization of the 5′-flanking region. J Biol
Chem. 264:2363–2367. 1989.PubMed/NCBI
|
|
38
|
Jandl JH, Inman JK, Simmons RL and Allen
DW: Transfer of iron from serum iron-binding protein to human
reticulocytes. J Clin Invest. 38:161–185. 1959. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Donovan A, Lima CA, Pinkus JL, Pinkus GS,
Zon LI, Robine S and Andrews NC: The iron exporter
ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab.
1:191–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Theil EC: Ferritin: Structure, gene
regulation, and cellular function in animals, plants, and
microorganisms. Annu Rev Biochem. 56:289–315. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ganz T: Hepcidin, a key regulator of iron
metabolism and mediator of anemia of inflammation. Blood.
102:783–788. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Berridge MV, Herst PM and Tan AS:
Tetrazolium dyes as tools in cell biology: New insights into their
cellular reduction. Biotechnol Annu Rev. 11:127–152. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Walter PB, Knutson MD, Paler-Martinez A,
Lee S, Xu Y, Viteri FE and Ames BN: Iron deficiency and iron excess
damage mitochondria and mitochondrial DNA in rats. Proc Natl Acad
Sci USA. 99:2264–2269. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ge XH, Wang Q, Qian ZM, Zhu L, Du F, Yung
WH, Yang L and Ke Y: The iron regulatory hormone hepcidin reduces
ferroportin 1 content and iron release in H9C2 cardiomyocytes. J
Nutr Biochem. 20:860–865. 2009. View Article : Google Scholar
|
|
46
|
Parkes JG, Liu Y, Sirna JB and Templeton
DM: Changes in gene expression with iron loading and chelation in
cardiac myocytes and non-myocytic fibroblasts. J Mol Cell Cardiol.
32:233–246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yoon D, Pastore YD, Divoky V, Liu E,
Mlodnicka AE, Rainey K, Ponka P, Semenza GL, Schumacher A and
Prchal JT: Hypoxia-inducible factor-1 deficiency results in
dysregulated erythropoiesis signaling and iron homeostasis in mouse
development. J Biol Chem. 281:25703–25711. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rolfs A, Kvietikova I, Gassmann M and
Wenger RH: Oxygen-regulated transferrin expression is mediated by
hypoxia-inducible factor-1. J Biol Chem. 272:20055–20062. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tacchini L, Bianchi L, Bernelli-Zazzera A
and Cairo G: Transferrin receptor induction by hypoxia.
HIF-1-mediated transcriptional activation and cell-specific
post-transcriptional regulation. J Biol Chem. 274:24142–24146.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Beaumont C: Molecular mechanisms of iron
homeostasis. Med Sci (Paris). 20:68–72. 2004. View Article : Google Scholar
|
|
51
|
Cairo G, Tacchini L, Pogliaghi G, Anzon E,
Tomasi A and Bernelli-Zazzer A: Induction of ferritin synthesis by
oxidative stress. Transcriptional and post-transcriptional
regulation by expansion of the 'free' iron pool. J Biol Chem.
270:700–703. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chandel NS, McClintock DS, Feliciano CE,
Wood TM, Melendez JA, Rodriguez AM and Schumacker PT: Reactive
oxygen species generated at mitochondrial complex III stabilize
hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2
sensing. J Biol Chem. 275:25130–25138. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Masso-Silva J, Diamond G, Macias-Rodriguez
M and Ascencio F: Genomic organization and tissue-specific
expression of hepcidin in the pacific mutton hamlet, Alphestes
immaculatus (Breder, 1936). Fish Shellfish Immunol. 31:1297–1302.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Merle U, Fein E, Gehrke SG, Stremmel W and
Kulaksiz H: The iron regulatory peptide hepcidin is expressed in
the heart and regulated by hypoxia and inflammation. Endocrinology.
148:2663–2668. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Przybylowski P, Malyszko J and Malyszko
JS: A possible role of hepcidin in the pathogenesis of anemia in
heart allograft recipients. Transplant Proc. 42:1803–1807. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Smith JJ, O'Brien-Ladner AR, Kaiser CR and
Wesselius LJ: Effects of hypoxia and nitric oxide on ferritin
content of alveolar cells. J Lab Clin Med. 141:309–317. 2003.
View Article : Google Scholar : PubMed/NCBI
|