Introduction
An orderly spread of action potentials through the
heart is critical for normal electrical function and its disruption
can lead to cardiac arrhythmias (1). Experiments in pre-clinical models
have advanced understanding of the electrophysiological mechanisms
underlying arrhythmogenesis using genetic and pharmacological
approaches (1–24). Experiments in mouse models have
highlighted the role of gap junctions in ventricular conduction and
arrhythmogenesis, however the results have been controversial.
Heterozygous Cx43+/− mice were demonstrated to exhibit
at 45–50% reduction in Cx43 expression, however, the degree of
conduction velocity (CV) slowing was variable: CV was either
unchanged (25–30) or reduced by 23–44% (31–33).
Additional experiments used a pharmacological approach,
demonstrating ventricular arrhythmogenesis associated with reduced
CV using 2 mM heptanol (7). This
agent inhibits gap junctions specifically at concentrations up to
1–2 mM (34,35) however at ≥2 mM additionally
inhibits sodium channels (34,36).
The extent to which the conduction defects and arrhythmogenesis
observed could be attributed to loss of gap junction coupling alone
remains to be fully elucidated.
Therefore, the aims of the present study were to
examine the possible role of abnormal gap junction function in
ventricular arrhythmogenesis, by applying heptanol at a low
concentration that specifically targets gap junctions (0.05 mM). At
this concentration, it was identified that heptanol did not elicit
spontaneous arrhythmias during regular pacing, however increased
the incidence of ventricular tachycardia induced by a S1S2
protocol. This was associated with increased activation latencies
in an absence of alterations in either action potential durations
(APDs) or effective refractory periods (ERPs). The observations of
the present study suggest that loss of gap junction function alone
is sufficient to produce conduction slowing and ventricular
arrhythmogenesis.
Materials and methods
Solutions
The experiments described in this study used
Krebs-Henseleit solution (composition in mM: NaCl 119,
NaHCO3 25, KCl 4, KH2PO4 1.2,
MgCl2 1, CaCl2 1.8, glucose 10 and sodium
pyruvate 2; pH 7.4) that had been bicarbonate-buffered and bubbled
with 95% O2/5% CO2 (37). Heptanol (0.82 g ml−1;
Sigma-Aldrich, Haverhill, UK) is soluble in aqueous solutions up to
9 mM (https://www.rsc.org/merck-index), and
was diluted using Krebs-Henseleit solution to produce a final
concentration of 0.05 mM.
Preparation of Langendorff-perfused
mouse hearts
A total of 13 wild-type mice of genetic background
129 (5 and 7 months of age, 3 male, 10 female; weight, 39.2±1.9 g)
were used in the current study. The animals were maintained at room
temperature (21±1°C) and were subjected to a 12:12 h light/dark
cycle with free access to sterile rodent chow and water in an
animal facility. The experiments described here were compliant with
the UK Animals (Scientific Procedures) Act 1986. The present study
was approved by the Animal Welfare and Ethical Review Body at
University of Cambridge. (Cambridge, UK) The procedures for the
preparation of Langendorff-perfused mouse hearts were as follows:
Mice were sacrificed by cervical dislocation in accordance with
Sections 1 (c) and 2 of Schedule 1 of the UK Animals (Scientific
Procedures) Act 1986. The hearts were rapidly excised and
immediately submerged in ice-cold Krebs-Henseleit solution.
Cannulation of the aorta was achieved using a tailor-made 21-gauge
cannula that had been prefilled with ice-cold buffer. Using a
micro-aneurysm clip (Harvard Apparatus, Cambridge, UK), the heart
was securely attached to the perfusion system. Retrograde perfusion
was initiated at a rate of 2–2.5 ml min−1 using a
peristaltic pump (Model 505S Bredel Pump; Watson-Marlow, Ltd.,
Falmouth, UK) with the perfusate passing through 200 and 5 µm
filters successively, and heated to 37°C using a water jacket and
circulator prior to reaching the aorta. The hearts that regained
their pink colour and spontaneous rhythmic activity were studied
further (approximately 90%). The remaining 10% were discarded.
Perfusion took place for a further 20 min to minimise any residual
effects of catecholamine released endogenously, prior to
electrophysiological analysis of the hearts.
Stimulation protocols
Electrical stimulation was achieved using paired
platinum electrodes (1 mm interpole distance) placed at the right
ventricular epicardium. This took place at 8 Hz, using square wave
pulses 2 msec in duration, with a stimulation voltage set to three
times the diastolic threshold (Grass S48 Square Pulse Stimulator;
Grass-Telefactor; Astro Med, Inc., Slough, UK) immediately
subsequent to the start of perfusion. The S1S2 protocol was used to
assess arrhythmogenicity and identify re-entrant substrates. This
consisted of a drive train of 8 regularly paced S1 stimuli
separated by a 125 msec basic cycle length (BCL), followed by
premature S2 extra stimuli every ninth stimulus. The S1S2 interval
was first set to 125 msec and then successively reduced by 1 msec
with each nine stimulus cycles until arrhythmic activity was
initiated or refractoriness was reached, whereupon the S2 stimulus
elicited no ventricular response.
Recording procedures
Monophasic action potential (MAP) recordings from
the left ventricular epicardium were obtained using a MAP electrode
(Linton Instruments; Harvard Apparatus). MAPs from the left
ventricular endocardium were obtained using a custom-made MAP
electrode that was made from two strands of 0.25 mm Teflon-coated
silver wire (99.99% purity; Advent Research Materials, Ltd.,
Witney, UK). To eliminate direct current offset, the electrode tips
were galvanically chlorided. The stimulating and recording
electrodes were maintained at constant positions, with an
inter-electrode distance of 3 mm. This allowed CVs to be determined
from the activation latencies. All recordings were performed using
a BCL of 125 msec (8 Hz) to exclude rate-dependent differences in
APDs. MAPs were pre-amplified using a NL100AK head stage, amplified
with a NL104A amplifier and band pass filtered between 0.5 Hz and 1
kHz using a NL125/6 filter (Neurolog; Digitimer, Ltd., Welwyn
Garden City, UK) and then digitized (1401plus MKII; Cambridge
Electronic Design, Ltd., Cambridge, UK) at 5 kHz. They were then
analysed using Spike2 version 5.11 software (Cambridge Electronic
Design, Ltd.). MAP waveforms that did not match the previous
established stringent criteria for MAP signals were rejected
(38). They must have stable
baselines, fast upstrokes, with no inflections or negative spikes,
and a rapid first phase of repolarization. 0% repolarization was
measured at the peak of the MAP and 100% repolarization was
measured at the point of return of the potential to baseline
(38–40).
The following parameters were obtained from the
experimental records: i) Activation latency, defined as the time
difference between the stimulus and the peak of the MAP; ii) CV, as
the ratio of the inter-electrode distance to the activation
latency. As the latter distance was kept constant, CVs were
inversely proportional to the corresponding activation latencies;
iii) APDx, the time difference between the peak of the
MAP and x=30, 50, 70 and 90% repolarization; iv) ERP, defined as
the longest S1S2 interval at which the S2 extra stimulus failed to
initiate a ventricular signal during programmed electrical
stimulation; v) excitation wavelength, λ, given by CV × ERP; vi)
critical intervals for re-excitation given by APD90 -
ERP.
Statistical analysis
All values are expressed as the mean ± standard
error. Categorical data were compared with Fisher's exact test
(one-tailed) using OriginPro version 8 (OriginLab Corporation,
Northampton, MA, USA). Different experimental groups were compared
by one-way analysis of variance. P<0.05 was considered to
indicate a statistically significant difference. P<0.05, 0.01
and 0.001 were denoted by *, ** and ***, respectively.
Results
Ventricular arrhythmogenicity and its association
with action potential activation and recovery properties were
examined prior and subsequent to introduction of 0.05 mM heptanol
in Langendorff-perfused mouse hearts. The right ventricular
epicardium was electrically stimulated using either regular 8 Hz or
S1S2 pacing (2,3,5,7,12).
MAP recordings were obtained from the left ventricular epicardium.
The stimulating and recording electrodes were maintained at a
constant distance of 3 mm, which permitted CVs to be estimated from
the respective activation latencies. Ventricular tachycardia (VT)
was defined as a series of ≥5 action potentials with coupling
intervals closer than the BCL.
Heptanol at 0.05 mM exerts ventricular
pro-arrhythmic effects during S1S2, however not during regular
pacing
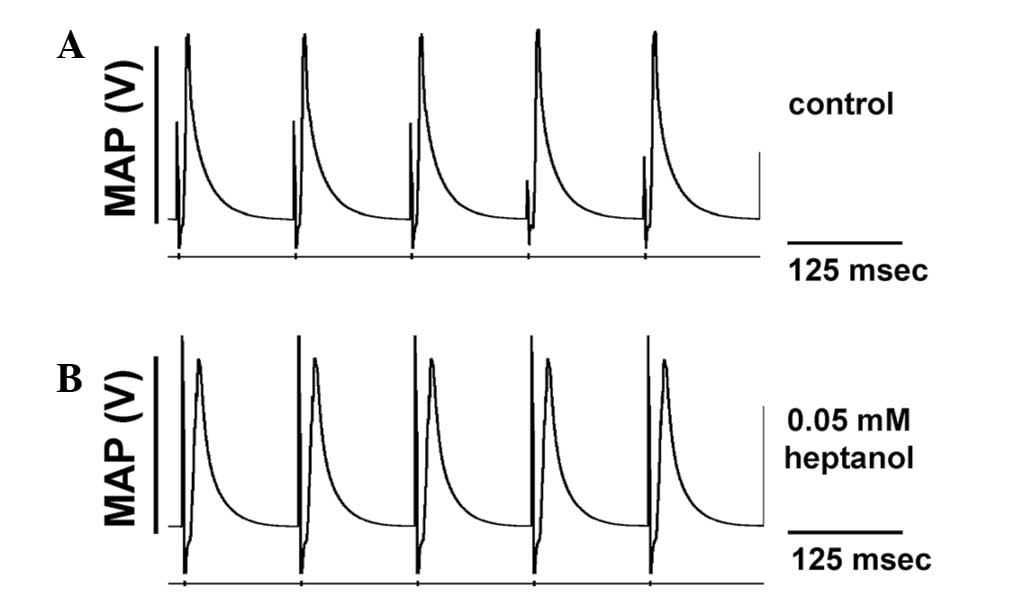
The initial experiments conducted during regular
pacing demonstrated consistent ventricular activity in the absence
of spontaneous arrhythmias in all of the 12 hearts studied, whether
prior or subsequent to introduction of 0.05 mM heptanol, or
following removal of heptanol from the perfusate (Fig. 1). The second set of experiments
then applied a S1S2 pacing protocol, which imposed extra systolic
S2 stimuli following trains of regular S1 pacing stimuli. The S1S2
interval was initially at the BCL and subsequently reduced by 1
msec with each cycle until the S2 stimuli produced either
arrhythmic activity or refractoriness. The latter indicating that
the ERP was reached. None of the hearts studied demonstrated
evidence of inducible arrhythmias prior to application of the test
agent (Fig. 2A). By contrast, it
was possible to induce VT subsequent to application of heptanol
(Fig. 2B). The incidences of
inducible VT prior and subsequent to introduction of heptanol, and
following its withdrawal from the perfusing solution are summarized
in Fig. 2C, indicating that
heptanol exerted significant pro-arrhythmic effects, as the extra
stimuli were able to induce VT in 6/12 hearts (*P<0.05; Fisher's
exact test).
Pro-arrhythmic effects of heptanol
were associated with reduced CVs in an absence of alterations in
APDs or ERPs
Previous studies in mouse models have associated
increased arrhythmogenicity with reduced CVs, prolonged or
shortened APDs and reduce ERPs (2,3,5,7,12).
These values were therefore obtained from the experimental
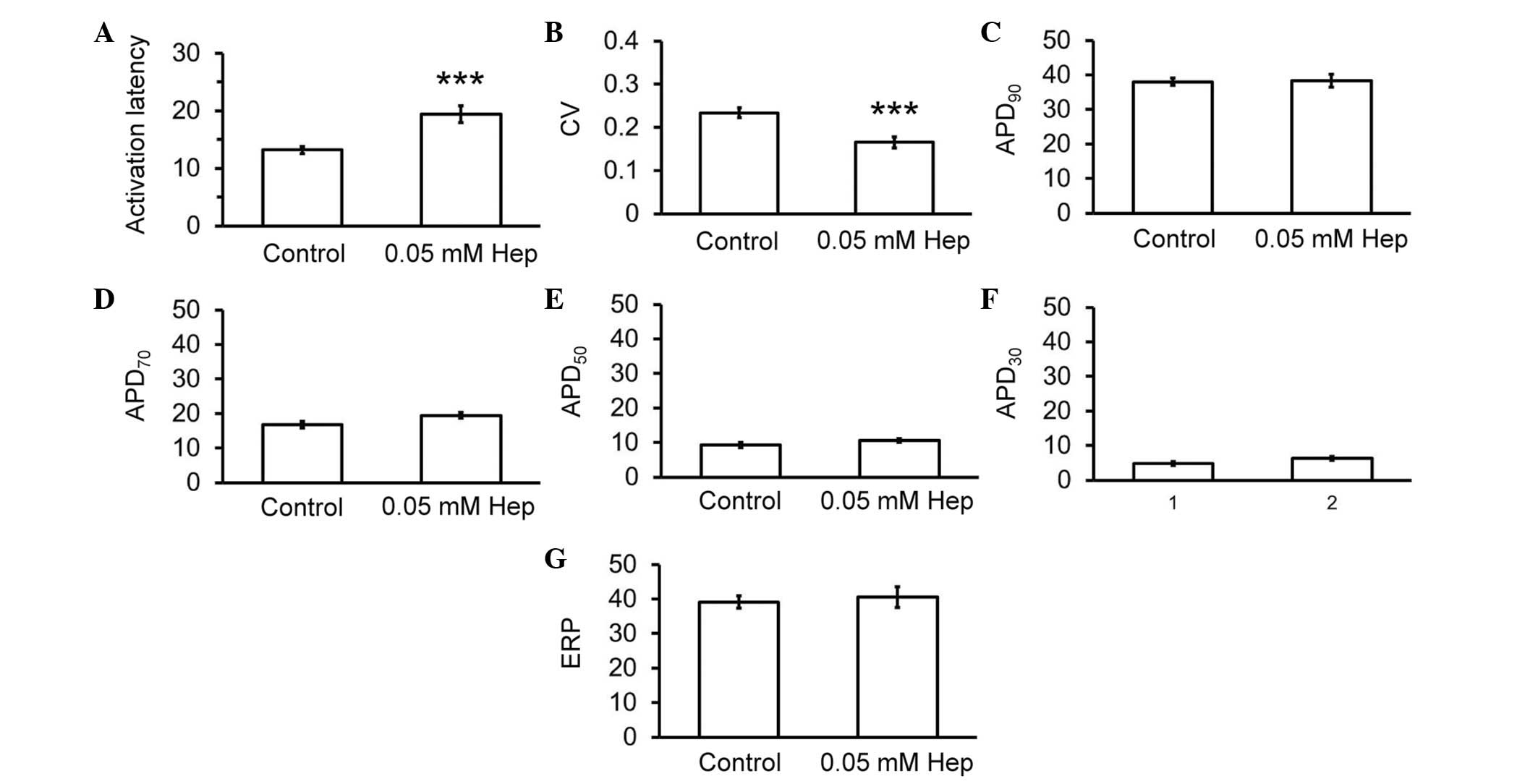
recordings described above. Thus, heptanol increased activation
latencies from 13.2±0.6 to 19.4±1.3 msec (Fig. 3A; analysis of variance; P<0.001)
and reduced CVs from 0.23±0.01 to 0.16±0.01 msec (Fig. 3B; P<0.001), without altering
APD90 (38.0±1.0 vs. 38.3±1.8 msec; Fig. 3C), APD70 (16.8±1.0 vs.
19.5±0.9 msec; Fig. 3D),
APD50 (9.2±0.8 vs. 10.1±0.6 msec; Fig. 3E), APD30 (4.8±0.5 vs.
6.3±0.6 msec; Fig. 3F) or ERPs
(39.6±1.9 vs. 40.6±3.0 msec; Fig.
3G).
Pro-arrhythmic effects of heptanol
were associated with reduced excitation wavelengths despite
unaltered critical intervals
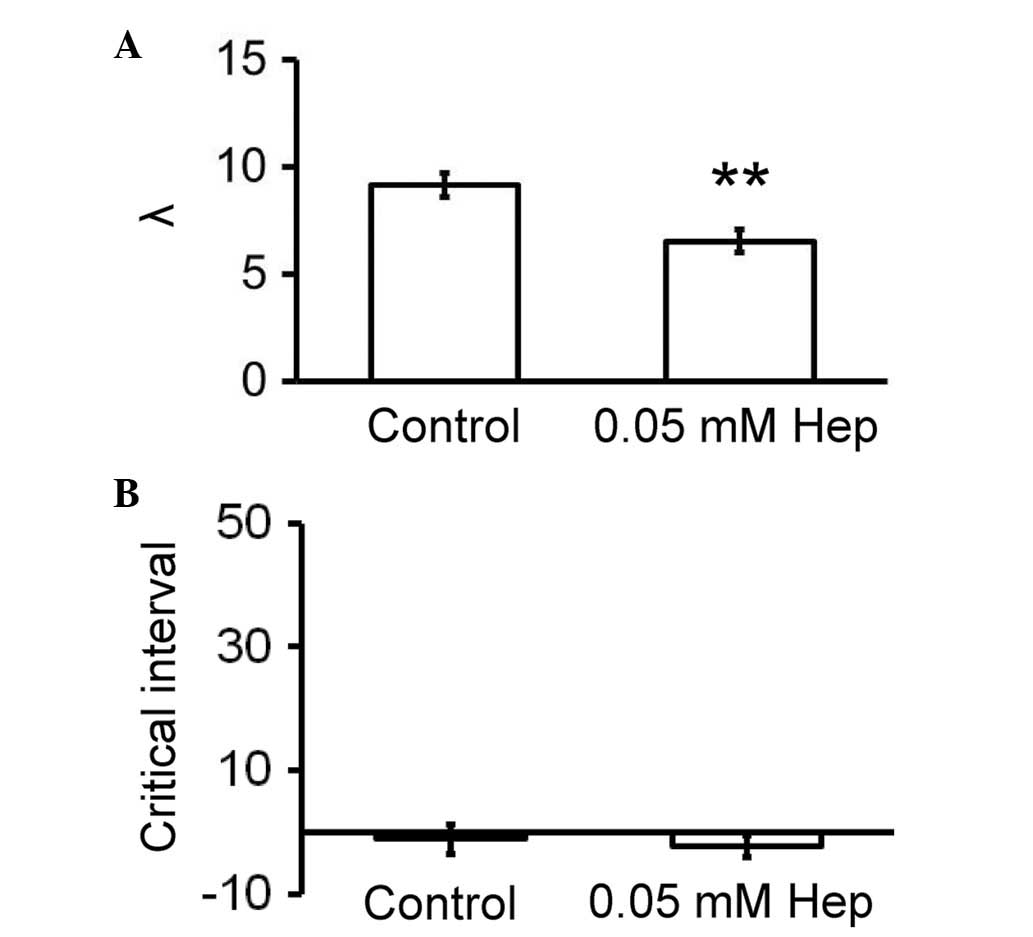
Reductions in excitation wavelengths (λ; CV × ERP)
and increases in critical intervals for re-excitation
(APD90 - ERP) have been associated with increased
arrhythmogenicity (7,41). Accordingly, these parameters were
calculated for the hearts used in the current study. Heptanol
reduced λ from 9.1±0.6 to 6.5±0.6 mm (Fig. 4A; P<0.01) without altering
critical intervals (−1.1±2.4 vs. −2.3±1.8 msec; Fig. 4B).
Discussion
Sudden cardiac death (SCD) is a significant problem
and is responsible for around 60,000 deaths in the UK (42), 200,000 deaths in the US (43) and 4–5 million deaths globally
(44) per year. It has been
suggested that SCD arises from the development of malignant
ventricular arrhythmias, the electrophysiological mechanisms of
which remain to be fully understood. Mouse hearts have been used to
study arrhythmogenesis as they are amenable to both genetic and
pharmacological manipulation.
Propagation of the action potentials through the
working myocardium depends on sodium channel activation followed by
gap junction conduction. Gap junctions are hexameric proteins made
of connexins mediate intercellular coupling by allowing passive
electrotonic spread of ions and of larger molecules (45). Their resistance contributes to
axial resistance and modulates CV (46,47).
Cx43 is the isoform present in ventricles, and the effects of loss
of Cx43 on ventricular conduction and arrhythmogenesis have been
extensively studied in mouse models (25–33,48,49),
however, with significant disagreement between the results of these
studies (28). Thus,
cardiac-restricted Cx43 inactivation followed by crossing with Cre
recombinase produced mosaic mice, in which Cx43 was observed to be
educed by up to 95% when compared with wild-type mice (48). Additional experiments identified
that heterozygous Cx43+/− mice exhibited a 45–50%
reduction in Cx43 expression. In these mice, CV was either
unchanged (25–30) or reduced by 23–44% (31–33).
These studies suggest different parameters, including interstitial
volume (50), width of the
perinexus, intracellular calcium concentrations, perfusate
composition and osmolarity (28),
have additional effects on cardiac conduction. Pharmacological
methods have additionally been used to study the role of gap
junctions in arrhythmogenesis. A previous study reported that 2 mM
heptanol exerted significant pro-arrhythmic effects by reducing CVs
without influencing APDs, however increased ERPs (7). These alterations led to reduced
excitation wavelength (λ; CV × ERP), which is consistent with the
increased likelihood of re-entry. Heptanol is an agent that
reversibly inhibits gap junctions at concentrations up to 1 mM and
also sodium channels at concentrations ≥2 mM (34,36).
It was therefore not possible to determine the relative
contributions of gap junction uncoupling vs. reduced sodium channel
function in the reduction of CV and the ventricular
arrhythmogenesis observed. Furthermore, 2 mM heptanol produced not
only CV slowing, however additionally increased ERPs. The latter
observation is consistent with its effects on sodium channel
kinetics of producing a depolarizing shift of the activation curve,
and a hyperpolarizing shift of the inactivation curve, which would
reduce the sodium window current (36). Increasing ERP alone is suggested to
be anti-arrhythmic via the increase in λ, regional increases in ERP
could theoretically predispose to re-entry by producing refractory
obstacles around which action potentials can circulate, and areas
of unidirectional conduction block (51).
Therefore, the present experiments were conducted to
determine whether heptanol at a concentration that specifically
inhibits gap junctions (0.05 mM) (34,36)
could produce pro-arrhythmic effects. Its application resulted in
an increased incidence of inducible, however not spontaneous,
arrhythmias, which was associated with increased activation
latencies and reduced CVs, in an absence of alterations in APDs or
ERPs. Together, these alterations led to a reduced excitation
wavelength (λ) despite leaving critical intervals unaltered. These
results are consistent with previous observations that inhibition
of gap junctions and sodium channels at 2 mM heptanol resulted in a
greater degree of CV slowing compared with the low concentration
used in the current study, and increased ERPs. Under these
conditions, spontaneous and provoked VT were observed. In the
present study, gap junction inhibition alone using 0.05 mM heptanol
did not elicit spontaneous VT during regular pacing.
As the aim of this study was to examine the effects
of reducing gap junction coupling, it was therefore appropriate to
use the MAP method. This method has been extensively used to study
cardiac electrophysiology in animal systems (8,52–59).
For future experiments, the measurement of magnetic signals may be
beneficial. It has been previously demonstrated to useful for
characterizing cardiac structural abnormalities (60–62),
and observed that functional mapping could be achieved using
magnetocardiography in mouse models. Thus, it is suggested that its
use in assessing abnormal cardiac electrophysiology in mice
warrants future investigation (63–66).
In conclusion, the current study demonstrated that
gap junction inhibition by heptanol alone was sufficient to reduce
CV without affecting APD or ERP, and the consequent reduction in λ
was suggested to be responsible for the arrhythmogenesis
observed.
Acknowledgements
Dr Gary Tse was awarded a BBSRC Doctoral Training
Award from the University of Cambridge (Cambridge, UK).
References
|
1
|
Tse G: Both transmural dispersion of
repolarization and transmural dispersion of refractoriness are poor
predictors of arrhythmogenicity: A role for the index of Cardiac
Electrophysiological Balance (QT/QRS)? J Geriatr Cardiol. 2016.
|
|
2
|
Tse G, Tse V and Yeo JM: Ventricular
anti-arrhythmic effects of heptanol in hypokalaemic,
Langendorff-perfused mouse hearts. Biomed Rep. 4:313–324.
2016.PubMed/NCBI
|
|
3
|
Tse G, Tse V, Yeo JM and Sun B: Atrial
anti-arrhythmic effects of heptanol in Langendorff-perfused mouse
hearts. PLoS One. 11:e01488582016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tse G: Mechanisms of cardiac arrhythmias.
J Arrhythm. 32:75–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tse G, Wong ST, Tse V and Yeo JM:
Restitution analysis of alternans using dynamic pacing and its
comparison with S1S2 restitution in heptanol-treated, hypokalaemic
Langendorff-perfused mouse hearts. Biomed Rep. 4:673–680.
2016.PubMed/NCBI
|
|
6
|
Tse G and Yeo JM: Conduction abnormalities
and ventricular arrhythmogenesis: The roles of sodium channels and
gap junctions. Int J Cardiol Heart Vasc. 9:75–82. 2015.PubMed/NCBI
|
|
7
|
Tse G, Hothi SS, Grace AA and Huang CL:
Ventricular arrhythmogenesis following slowed conduction in
heptanol-treated, Langendorff-perfused mouse hearts. J Physiol Sci.
62:79–92. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tse G, Wong ST, Tse V and Yeo JM:
Monophasic action potential recordings: Which is the recording
electrode? J Basic Clin Physiol Pharmacol. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tse G, Lai TH, Yeo JM, Tse V and Wong SH:
Mechanisms of electrical activation and conduction in the
gastrointestinal system: Lessons from cardiac electrophysiology.
Front Physiol. 7:1822016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tse G, Wong ST, Tse V and Yeo JM:
Depolarization vs. repolarization: What is the mechanism of
ventricular arrhythmogenesis underlying sodium channel
haploinsufficiency in mouse hearts? Acta Physiol (Oxf). 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Z, Sun B, Tse G, Jiang J and Xu W:
Reversibility of both sinus node dysfunction and reduced HCN4 mRNA
expression level in an atrial tachycardia pacing model of
tachycardia-bradycardia syndrome in rabbit hearts. Int J Clin Exp
Pathol. 9:2016.
|
|
12
|
Tse G, Sun B, Wong ST, Tse V and Yeo JM:
Ventricular anti-arrhythmic effects of hypercalcaemia treatment in
hyperkalaemic, Langendorff-perfused mouse hearts. Biomed Rep.
4:313–324. 2016.PubMed/NCBI
|
|
13
|
Tse G, Lai ET, Tse V and Yeo JM: Molecular
and electrophysiological mechanisms underlying cardiac
arrhythmogenesis in diabetes mellitus. J Diabetes Res. 2016.(In
press). View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tse G, Wong ST, Tse V and Yeo JM:
Determination of action potential wavelength restitution in
Scn5a+/− mouse hearts modelling human Brugada syndrome. J Physiol.
2016.(In press).
|
|
15
|
Osadchii OE: Flecainide-induced
proarrhythmia is attributed to abnormal changes in repolarization
and refractoriness in perfused guinea-pig heart. J Cardiovasc
Pharmacol. 60:456–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Osadchii OE: Quinidine elicits
proarrhythmic changes in ventricular repolarization and
refractoriness in guinea-pig. Can J Physiol Pharmacol. 91:306–315.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilde AA, Postema PG, Di Diego JM, Viskin
S, Morita H, Fish JM and Antzelevitch C: The pathophysiological
mechanism underlying Brugada syndrome: Depolarization versus
repolarization. J Mol Cell Cardiol. 49:543–553. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Osadchii OE: Impact of hypokalemia on
electromechanical window, excitation wavelength and repolarization
gradients in guinea-pig and rabbit hearts. PLoS One. 9:e1055992014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Osadchii OE: Impaired epicardial
activation-repolarization coupling contributes to the proarrhythmic
effects of hypokalaemia and dofetilide in guinea pig ventricles.
Acta Physiol (Oxf). 211:48–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsieh YC, Lin JC, Hung CY, Li CH, Lin SF,
Yeh HI, Huang JL, Lo CP, Haugan K, Larsen BD and Wu TJ: Gap
junction modifier rotigaptide decreases the susceptibility to
ventricular arrhythmia by enhancing conduction velocity and
suppressing discordant alternans during therapeutic hypothermia in
isolated rabbit hearts. Heart Rhythm. 13:251–261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsieh YC, Lin SF, Huang JL, Hung CY, Lin
JC, Liao YC, Lo CP, Wang KY and Wu TJ: Moderate hypothermia (33°C)
decreases the susceptibility to pacing-induced ventricular
fibrillation compared with severe hypothermia (30°C) by attenuating
spatially discordant alternans in isolated rabbit hearts. Zhonghua
Minguo Xin Zang Xue Hui Za Zhi. 30:455–465. 2014.
|
|
22
|
Hsieh YC, Lin SF, Lin TC, Ting CT and Wu
TJ: Therapeutic hypothermia (30 degrees C) enhances arrhythmogenic
substrates, including spatially discordant alternans, and
facilitates pacing-induced ventricular fibrillation in isolated
rabbit hearts. Circ J. 73:2214–2222. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choy L, Yeo JM, Tse V, Chan SP and Tse G:
Cardiac disease and arrhythmogenesis: Mechanistic insights from
mouse studies. Int J Cardiol Heart Vasc. 12:1–10. 2016.PubMed/NCBI
|
|
24
|
Tse G, Lai ET, Chan YWF, Yeo JM and Yan
BP: What is the arrhythmic substrate in viral myocarditis? Insights
from clinical and animal studies. Front Physiol. 7:3082016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stein M, van Veen TA, Remme CA, Boulaksil
M, Noorman M, van Stuijvenberg L, van der Nagel R, Bezzina CR,
Hauer RN, de Bakker JM and van Rijen HV: Combined reduction of
intercellular coupling and membrane excitability differentially
affects transverse and longitudinal cardiac conduction. Cardiovasc
Res. 83:52–60. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Stein M, van Veen TA, Hauer RN, de Bakker
JM and van Rijen HV: A 50% reduction of excitability but not of
intercellular coupling affects conduction velocity restitution and
activation delay in the mouse heart. PLoS One. 6:e203102011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morley GE, Vaidya D, Samie FH, Lo C,
Delmar M and Jalife J: Characterization of conduction in the
ventricles of normal and heterozygous Cx43 knockout mice using
optical mapping. J Cardiovasc Electrophysiol. 10:1361–1375. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
George SA, Sciuto KJ, Lin J, Salama ME,
Keener JP, Gourdie RG and Poelzing S: Extracellular sodium and
potassium levels modulate cardiac conduction in mice heterozygous
null for the Connexin43 gene. Pflugers Arch. 467:2287–2297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vaidya D, Tamaddon HS, Lo CW, Taffet SM,
Delmar M, Morley GE and Jalife J: Null mutation of connexin43
causes slow propagation of ventricular activation in the late
stages of mouse embryonic development. Circ Res. 88:1196–1202.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Rijen HV, Eckardt D, Degen J, Theis M,
Ott T, Willecke K, Jongsma HJ, Opthof T and de Bakker JM: Slow
conduction and enhanced anisotropy increase the propensity for
ventricular tachyarrhythmias in adult mice with induced deletion of
connexin43. Circulation. 109:1048–1055. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guerrero PA, Schuessler RB, Davis LM,
Beyer EC, Johnson CM, Yamada KA and Saffitz JE: Slow ventricular
conduction in mice heterozygous for a connexin43 null mutation. J
Clin Invest. 99:1991–1998. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomas SA, Schuessler RB, Berul CI,
Beardslee MA, Beyer EC, Mendelsohn ME and Saffitz JE: Disparate
effects of deficient expression of connexin43 on atrial and
ventricular conduction: Evidence for chamber-specific molecular
determinants of conduction. Circulation. 97:686–691. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eloff BC, Lerner DL, Yamada KA, Schuessler
RB, Saffitz JE and Rosenbaum DS: High resolution optical mapping
reveals conduction slowing in connexin43 deficient mice. Cardiovasc
Res. 51:681–690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Christ GJ, Spektor M, Brink PR and Barr L:
Further evidence for the selective disruption of intercellular
communication by heptanol. Am J Physiol. 276:H1911–H1917.
1999.PubMed/NCBI
|
|
35
|
Rüdisüli A and Weingart R: Electrical
properties of gap junction channels in guinea-pig ventricular cell
pairs revealed by exposure to heptanol. Pflugers Arch. 415:12–21.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nelson WL and Makielski JC: Block of
sodium current by heptanol in voltage-clamped canine cardiac
Purkinje cells. Circ Res. 68:977–983. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Balasubramaniam R, Grace AA, Saumarez RC,
Vandenberg JI and Huang CL: Electrogram prolongation and
nifedipine-suppressible ventricular arrhythmias in mice following
targeted disruption of KCNE1. J Physiol. 552:535–546. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Knollmann BC, Katchman AN and Franz MR:
Monophasic action potential recordings from intact mouse heart:
Validation, regional heterogeneity, and relation to refractoriness.
J Cardiovasc Electrophysiol. 12:1286–1294. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gussak I, Chaitman BR, Kopecky SL and
Nerbonne JM: Rapid ventricular repolarization in rodents:
Electrocardiographic manifestations, molecular mechanisms, and
clinical insights. J Electrocardiol. 33:159–170. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fabritz L, Kirchhof P, Franz MR, Eckardt
L, Mönnig G, Milberg P, Breithardt G and Haverkamp W: Prolonged
action potential durations, increased dispersion of repolarization,
and polymorphic ventricular tachycardia in a mouse model of
proarrhythmia. Basic Res Cardiol. 98:25–32. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wiener N and Rosenblueth A: The
mathematical formulation of the problem of conduction of impulses
in a network of connected excitable elements, specifically in
cardiac muscle. Arch Inst Cardiol Mex. 16:205–265. 1946.PubMed/NCBI
|
|
42
|
Implantable cardioverter defibrillators
for arrhythmias, . Review of technology appraisal 11. National
Institute for Health and Clinical Excellence (NICE); 2007
|
|
43
|
Adabag AS, Luepker RV, Roger VL and Gersh
BJ: Sudden cardiac death: Epidemiology and risk factors. Nat Rev
Cardiol. 7:216–225. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chugh SS, Reinier K, Teodorescu C, Evanado
A, Kehr E, Al Samara M, Mariani R, Gunson K and Jui J: Epidemiology
of sudden cardiac death: Clinical and research implications. Prog
Cardiovasc Dis. 51:213–228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Spray DC and Burt JM: Structure-activity
relations of the cardiac gap junction channel. Am J Physiol.
258:C195–C205. 1990.PubMed/NCBI
|
|
46
|
Dhillon PS, Gray R, Kojodjojo P, Jabr R,
Chowdhury R, Fry CH and Peters NS: Relationship between
gap-junctional conductance and conduction velocity in mammalian
myocardium. Circ Arrhythm Electrophysiol. 6:1208–1214. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Peters NS: Gap junctions: Clarifying the
complexities of connexins and conduction. Circ Res. 99:1156–1158.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gutstein DE, Morley GE, Tamaddon H, Vaidya
D, Schneider MD, Chen J, Chien KR, Stuhlmann H and Fishman GI:
Conduction slowing and sudden arrhythmic death in mice with
cardiac-restricted inactivation of connexin43. Circ Res.
88:333–339. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Beauchamp P, Choby C, Desplantez T, de
Peyer K, Green K, Yamada KA, Weingart R, Saffitz JE and Kléber AG:
Electrical propagation in synthetic ventricular myocyte strands
from germline connexin43 knockout mice. Circ Res. 95:170–178. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Veeraraghavan R, Salama ME and Poelzing S:
Interstitial volume modulates the conduction velocity-gap junction
relationship. Am J Physiol Heart Circ Physiol. 302:H278–H286. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hondeghem LM: QTc prolongation as a
surrogate for drug-induced arrhythmias: Fact or fallacy? Acta
Cardiol. 66:685–689. 2011.PubMed/NCBI
|
|
52
|
Vigmond EJ: The electrophysiological basis
of MAP recordings. Cardiovasc Res. 68:502–503. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vigmond EJ and Leon LJ:
Electrophysiological basis of mono-phasic action potential
recordings. Med Biol Eng Comput. 37:359–365. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tse G: Novel conduction-repolarization
indices for the stratification of arrhythmic risk. J Geriatr
Cardiol. 2016.(Accepted).
|
|
55
|
Tse G: (Tpeak-Tend)/QRS and
(Tpeak-Tend)/(QT x QRS): Novel markers for predicting arrhythmic
risk in Brugada syndrome. Europace. 2016.(Accepted). View Article : Google Scholar
|
|
56
|
Tse G and Yan BP: Novel arrhythmic risk
markers incorporating QRS dispersion: QRSd x (Tpeak-Tend)/QRS and
QRSd x (Tpeak-Tend)/(QT x QRS). Ann Noninvasive Electrocardiol.
2016.(Accepted). View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tse G, Lai ET, Yeo JM and Yan BP:
Electrophysiological mechanisms of Bayés syndrome: Insights from
clinical and mouse studies. Front Physiol. 7:1882016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tse G, Lai ET, Lee AP, Yan BP and Wong SH:
Electrophysiological mechanisms of gastrointestinal
arrhythmogenesis: Lessons from the heart. Front Physiol. 7:2016.
View Article : Google Scholar
|
|
59
|
Tse G, Wong ST, Tse V and Yeo JM:
Variability in local action potential durations, dispersion of
repolarization and wavelength restitution in aged wild-type and
Scn5a+/− mouse hearts modelling human Brugada syndrom. J Geriatr
Cardiol. 2016.(Accepted).
|
|
60
|
Tse G, Ali A, Alpendurada F, Prasad S,
Raphael CE and Vassiliou V: Tuberculous constrictive pericarditis.
Res Cardiovasc Med. 4:e296142015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tse G, Ali A, Prasad SK, Vassiliou V and
Raphael CE: Atypical case of post-partum cardiomyopathy: An overlap
syndrome with arrhythmogenic right ventricular cardiomyopathy?
BJR|case reports. 1:201501822015. View Article : Google Scholar
|
|
62
|
Vassiliou V, Chin C, Perperoglou A, Tse G,
Ali A, Raphael C, Jabbour A, Newby D, Pennell D, Dweck D and Prasad
S: 93 ejection fraction by cardiovascular magnetic resonance
predicts adverse outcomes post aortic valve replacement. Heart.
100:A53–A54. 2014. View Article : Google Scholar
|
|
63
|
Ono Y and Ishiyama A: Non-invasive cardiac
functional mapping on disease-model mice-development of high
spatial resolution SQUID system for MCG on mice. Teion Kogaku.
40:44–50. 2005. View Article : Google Scholar
|
|
64
|
Tse G and Yan BP: Traditional and novel
ECG conduction and repolarization markers of sudden cardiac death.
Europace. 2016.(In press).
|
|
65
|
Tse G, Yan BP, Chan YW, Tian XY and Huang
Y: Reactive oxygen species, endoplasmic reticulum stress and
mitochondrial dysfunction: the link with cardiac arrhythmogenesis.
Front Physiol. 7:3132016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hu Z, Chen Z, Wang Y, Jiang J, Tse G, Xu
W, Ge J and Sun B: Effects of granulocyte colony-stimulating factor
on rabbit carotid and swine heart models of chronic obliterative
arterial disease. Mol Med Rep. 2016.(Accepted).
|