Introduction
Nagashima-type palmoplantar keratosis (NPPK; OMIM
#615598) is an autosomal recessive form of palmoplantar keratoderma
(PPK), which exhibits a relatively high incidence and has only been
reported in Japanese and Chinese populations (1,2). The
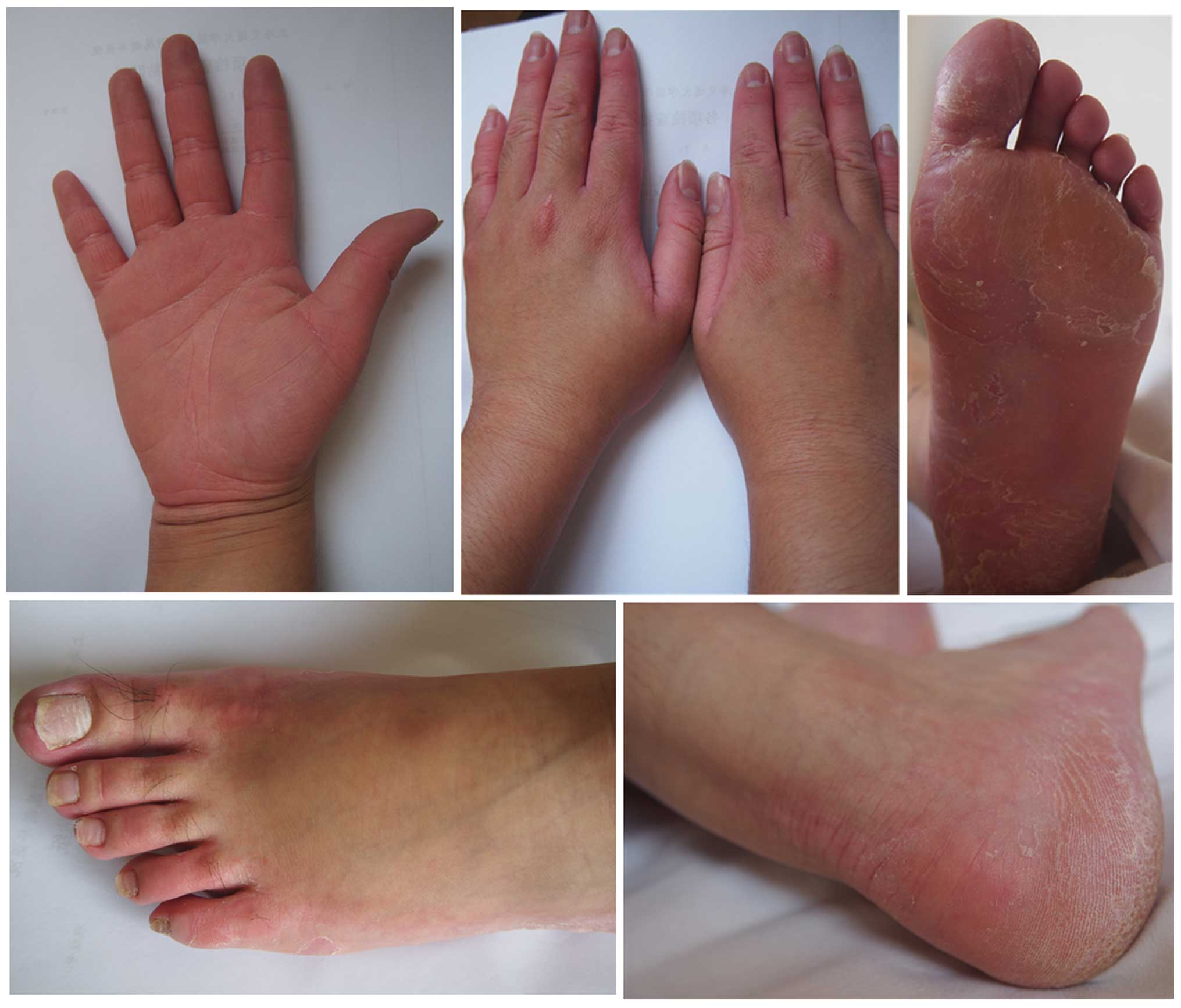
characteristic features of NPPK are erythema and hyperkeratosis of
the palms and soles, with sharp demarcation that mainly extends to
the wrists, ankles, Achilles tendon area, and the dorsal aspects of
the fingers and toes (1).
Furthermore, other frictional regions, such as knees and elbows,
may be involved. Some patients with NPPK also exhibit associated
palmoplantar features, including a white, spongy appearance within
10 min of water exposure, hyperhidrosis and fungal infections
(3).
The clinical features of diffuse PPK and the
recessive mode of inheritance are not unique characteristics for
NPPK, but are also associated with autosomal recessive exfoliative
ichthyosis (AREI; OMIM #607936), which is caused by mutations in
cystatin A (CSTA) (4), and
an atypical mild form of Mal de Meleda (MDM; OMIM #248300), which
is caused by mutations in secreted LY6/PLAUR domain containing 1
(SLURP1) (5). Other types
of autosomal dominant diffuse PPK with de novo mutations
include Unna-Thost type PPK [OMIM #600962; corresponding gene,
keratin (KRT)1], Vorner type PPK (OMIM #144200;
corresponding gene, KRT9/KRT1) and Bothnian type PPK
[OMIM 600231; corresponding gene, aquaporin 5 (AQP5)], which
must be differentiated from NPPK (3).
Mutations in the serpin peptidase inhibitor, clade B
(ovalbumin), member 70 (SERPINB7) gene, which encodes a
member of the serine protease inhibitor superfamily, results in a
complete loss of protease inhibitory activity. SERPINB7
mutations were reported to be responsible for NPPK in 2013
(3).
At present, only 31 unrelated, molecularly diagnosed
cases of NPPK associated with seven distinct pathogenic
SERPINB7 mutations in the homozygous or compound
heterozygous state have been reported in the literature. These
cases include the most popular founder mutation c.796C>T, and
other potentially frequent mutations c.218_219del2ins12,
c.336+2T>G, c.455-1G>A, c.455G>T, c.522dupT and
c.650_653delCTGT (2,3,6,7).
Among these studies, only one report regarding seven cases of NPPK
associated with four different SERPINB7 mutations was
available in Mainland China (2).
The present study investigated 12 suspected Chinese
patients with NPPK. Nine of the participants were definitely
diagnosed with NPPK by molecular analysis.
Materials and methods
Subjects
The present study was approved by the Institutional
Review Board of Xinhua Hospital, Shanghai Jiaotong University
School of Medicine (Shanghai, China). All participants provided
written informed consent. Patients suspected as having NPPK were
recruited by experienced dermatologists from Xinhua Hospital and
Shanghai Skin Hospital (Shanghai, China). In total, 12 probands (8
females and 4 males), nearly all of their parents (except for the
parents of Patients 11 and 12, and the father of Patient 10), and
100 population-matched healthy controls were enrolled in the
present study between August 2006 and December 2014. All probands
were genetically unrelated ethnic Han Chinese, which exhibited
non-progressive, symmetrical, diffuse erythema and hyperkeratosis
over the palms and soles from infancy. Elbow and knee involvement
was not observed. The clinical appearance of three patients
(Patients 1, 3 and 10; 8 months old, 19 years old and 22 years old,
respectively) is presented in Figs.
1–3. Clinical details were
available for 10 of the 12 patients. The age group ranged between 8
months and 51 years. The age of onset was ~3 months (Table I). No family history and
consanguinity was identified in this cohort.
 | Table I.Identified SERPINB7 mutations in the
present cohort. |
Table I.
Identified SERPINB7 mutations in the
present cohort.
|
|
|
| Molecular
results |
|---|
|
|
|
|
|
|---|
|
|
|
| Allele 1 | Allele 2 |
|---|
|
|
|
|
|
|
|---|
| Patient no. | Gender/age | Age at first
symptom | Nucleotide
change | Amino acid
change | Nucleotide
change | Amino acid
change |
|---|
| 1 | M/8 m | 2 m | c.796C>T | p.R266* | c.796C>T | p.R266* |
| 2 | F/2 y | 1 w | c.796C>T | p.R266* | c.522dupT | p.Val175fs |
| 3 | F/19 y | 3 m | c.796C>T | p.R266* | c.796C>T | p.R266* |
| 4 | M/4 y | 6 m | c.796C>T | p.R266* | c.796C>T | p.R266* |
| 5 | M/16 y | 3 m | c.796C>T | p.R266* | ? | ? |
| 6 | F/26 y | 2 m | c.796C>T | p.R266* | c.796C>T | p.R266* |
| 7 | F/17 y | 2 m | c.796C>T | p.R266* | ? | ? |
| 8 | F/24 y | 5 m | c.796C>T | p.R266* | ? | ? |
| 9 | F/36 y | ? | c.796C>T | p.R266* | c.796C>T | p.R266* |
| 10 | M/22 y | 3 m | c.796C>T | p.R266* |
c.122_127delTGGTCC | p.Leu41fs |
| 11 | F/51 y | ? | c.796C>T | p.R266* | c.522dupT | p.Val175fs |
| 12 | F/2 y | 2 m | c.796C>T | p.R266* | c.455G>T | Predicted splicing
alteration |
Methods
Peripheral blood samples were collected from all
participants for DNA extraction. DNA was extracted using a TIANamp
Blood DNA kit (Tiangen Biotech Co., Ltd., Beijing, China) and PCR
primers were designed flanking all coding exons and intron-exon
boundaries of six genes (SERPINB7, SLURP1,
AQP5, CSTA, KRT1 and KRT9) using Primer
Premier 5.0 software (Premier Biosoft, Palo Alto, CA, USA; Table II). Genomic DNA samples were
amplified by standard polymerase chain reaction and the PCR
protocol was as follows: i) Denaturation, 94°C for 5 min; ii) 31
cycles of denaturation at 94°C for 30 sec, annealing for 30 sec at
temperatures according to the primers for each fragment, and
extension at 72°C for 1 min; iii) extension, 72°C for 1 min; and
iv) extension, 4°C for 5 min. PCR was repeated 10–20 times. Sanger
sequencing was performed using an ABI PRISM®3730
automated sequencer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The SERPINB7 gene, which is
associated with autosomal recessive NPPK, was tested initially. If
no corresponding mutations in two alleles were identified, the
other five genes were sequentially sequenced. Identified mutations
were respectively confirmed in the unaffected family members (if
available) and 100 population-matched healthy controls (if mutation
is novel).
| Table II.Primers of the six screening genes
(SERPINB7, SLURP1, CSTA, AQP5, KRT1 and KRT9). |
Table II.
Primers of the six screening genes
(SERPINB7, SLURP1, CSTA, AQP5, KRT1 and KRT9).
|
| Primer sequence |
|
|---|
|
|
|
|
|---|
| Primer name | Forward | Reverse | Primer size (bp) |
|---|
| SERPINB7-E02 |
CAGAAATGTCCACCAACGAG |
ATATTTCTGCTGCCTCTTGG | 608 |
| SERPINB7-E03 |
CTTTCCTTGTGCCCTGTTTA |
TTAAGCTAACCTCCCACCAT | 295 |
| SERPINB7-E04 |
GGGCAAGAAAGGATGAAGTT |
CATCCCTACCAATAGACACG | 704 |
| SERPINB7-E05 |
CCTTCCAGTCCCATTTCCAT |
GAGGGTGAGATATTGAGGTT | 615 |
| SERPINB7-E06 |
CACAGGGATTATGTAAGGAT |
ACACGTTTGGTGGTGTTTCA | 559 |
| SERPINB7-E07 |
ACCCAAGGTCACATAGTTAG |
CTAGTATCTCAATACCCTGA | 485 |
| SERPINB7-E08 |
TCACCTGTCTATTGCTCCAC |
ATTGACTTGTGGTGGTTCTT | 765 |
| SLUPR1-E01 |
CAGAGGCACAGCCAGGACAT |
TAGGAGGTGGGCAGACAAGC | 470 |
| SLUPR1-E02 |
TCTGTGGCTCAGCTCAGTTAGA |
TCCCTGTTCCCAATAGTCCA | 709 |
| SLUPR1-E03 |
TGGACTATTGGGAACAGGGATC |
GGTTCAGAGTTCCGAGTTGC | 257 |
| CSTA-E01 |
TAAAACACGAGTCTCCACACT |
AAAGCCACAAACATCCTAAA | 256 |
| CSTA-E02 |
ACTTTTAGGAGGATGAGGTT |
AAGGAATTATGTGGTAGGGA | 284 |
| CSTA-E03_NEW |
ACCCATTTGAATGAATCTCC |
CCAGTTGCATTAGGCTTGAC | 433 |
| AQP5-E01 |
CGCCGCATCCACCTCCTCCG |
CCCCAGGGTCGAGGCTCCCA | 486 |
| AQP5-E02 |
AAAAGCCCTACTCCCCGAGC |
GATTCCTGTCCCATCCCACC | 466 |
| AQP5-E03 |
CAGGAATCAAACCCAACCTC |
TCCCTTTCTCTGTCAGCCAC | 442 |
| AQP5-E04 |
CGCTCTGTTCATCCGTCTCT |
TTTCTTCTTTTCCCCCTTGG | 576 |
| KRT1-E01A |
CCAAGCCCAATTTCTTCCCTG |
AAGGCTCTGGTTGATAGTGA | 549 |
| KRT1-E01B |
TGGAAGTCGGAGTCTTGTTA |
ATTCAACAGATATGAGTCCC | 696 |
| KRT1-E02 |
GTATGCGCTTTGCTATTGGT |
ATTGCCTATCACTGCCTTTC | 684 |
| KRT1-E03 |
TTAGGTTAGAGGCACATCAG |
AAATGTGAGTTCCGTCCTAC | 313 |
| KRT1-E04+05 |
CCATATTTCCCAGCACCTTA |
AGATGGTAGATAGCGTTTGT | 794 |
| KRT1-E06 |
CAAGGTGAGTGGGCTGAAAG |
CTCACATTGACCATCCCATC | 492 |
| KRT1-E07 |
AGTCTGTAAGGGTTGTAGGAG |
GAATAATTTGCTCCACCTCA | 699 |
| KRT1-E08+09 |
GCGGTTTGGGAAGCTGGAGT |
TTGAAATGTCATGTGGGTGG | 877 |
| KRT9-E01_in |
CGGTAGCACTCCTATCACTGC |
CTGCTCTGCCCAAACTCTGAA | 931 |
| KRT9-E02+03 |
ATCTTCGCTGAAGGCTGGAA |
AAGCCAAAGCCCAACCACTA | 701 |
| KRT9-E04 |
GTGGTTGGGCTTTGGCTTCA |
GGAGGTGGGAGGGATGGAGA | 356 |
| KRT9-E05+06 |
GACTTGTCATTGGCTTCAGA |
CAGAGGGACAGAAGTAGTATCA | 664 |
| KRT9-E07 |
AGATTCATGTTTGGGTCCTG |
CCCTTACCTTTTGTCTCATCT | 621 |
Results
Mutations identified in SERPINB7 (GenBank
accession number: NM_001040147.2) are summarized in Table I. The present study detected five
homozygous founder mutations (c.796C>T) and four compound
heterozygous mutations (c.796C>T combined with c.455G>T,
c.522dupT or c.122_127delTGGTCC) in nine patients with NPPK
(Fig. 4). Among the mutations, the
in-frame deletion mutation c.122_127delTGGTCC has not previously
been reported, and was also absent in the 100 population-matched
healthy controls. Furthermore, indexed unaffected parents in the
present cohort were all heterozygous carriers. Notably, the other
three patients were revealed to harbor one heterozygous founder
mutation (c.796C>T) in SERPINB7, whereas no pathogenic
mutations were detected in the five remaining candidate genes.
Discussion
All of the seven previously reported pathogenic
mutations associated with NPPK (c.218_219del2ins12, c.336+2T>G,
c.455-1G>A, c.455G>T, c.522dupT, c.650_653delCTGT,
c.796C>T) are nonsense/frameshift/splice site mutations, which
form premature stop codons and truncate the protein, thus
suggesting that missense variants without splicing effects tend to
be non-pathogenic polymorphisms. The novel mutation
c.122_127delTGGTCC (p.Leu41_Val42del) identified in the present
cohort was estimated to cause an in-frame deletion of two amino
acid residues (leucine and valine). Considering patient 10
exhibited the typical features of NPPK (Fig. 3), and the in-frame deletion
mutation was not detected in 200 alleles from the 100 healthy
controls, it was predicted that this mutation may shorten the
protein, and exert pathogenic effects resulting in an NPPK
phenotype.
Notably, three suspected patients with typical
palmoplantar lesions of NPPK were all shown to harbor one
heterozygous founder mutation (c.796C>T) in SERPINB7,
whereas no pathogenic mutations were identified in the remaining
five candidate genes responsible for analogous genodermatoses. It
has previously been reported that female carriers with missense
SLURP1 mutations may exhibit mild palmar lesions (8), whereas a heterozygous SERPINB7
mutation did not induce any palmoplantar abnormalities, thus
indicating that either of the alleles can retain protein activity
of SERPINB7 and sustain normal skin structure. Therefore, the three
suspected cases very probably suffer from NPPK. In addition, novel
mutations in the other allele may be undetectable large deletions,
or mutations located in other loci of SERPINB7, including
deep introns and 5′/3′-untranslated regions, which require more
intensive investigations, such as RNA testing or next-generation
sequencing. Unfortunately, these samples are not available for
retesting.
The major etiological factor associated with MDM is
consanguineous marriage (9,10),
and MDM has been diagnosed in the Mediterranean region, the Middle
East, Europe and East Asia. Conversely, NPPK appears to be an
Asian-only endemic (11), the
prevalence rate of which has been estimated to be 1.2/10,000 and
3.1/10,000 in Japanese and Chinese Han populations, respectively
(3). Furthermore, the most common
mutation (c.796C>T) exerts a founder effect and contributes to
NPPK etiology (2).
In order to detect other SERPINB7 variants
with a worldwide range that harbor the potential for being
pathogenic, they were searched for using the 1000 Genomes Browser
Version 3.0.2 (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/).
The corresponding results are summarized in Table III. Furthermore, in combination
with previous studies, the c.218_219del2ins12 mutation has only
been detected in the Japanese population; the c.522dupT and
c.650_653delCTGT mutations were only detected in the Chinese Han
population; and four known pathogenic mutations (c.336+2T>G,
c.455-1G>A, c.455G>T and c.796C>T) existed in both
populations (2,3,7).
Notably, c.455G>T and c.522dupT have been reported in two
unrelated patients, and the c.455G>T mutation has a high allele
frequency of 0.0025, in the Chinese Han population, thus suggesting
a potential founder effect (7).
| Table III.Allele frequencies of known
pathogenic mutations (in bold) and other potential pathogenic
variants in distinct population. |
Table III.
Allele frequencies of known
pathogenic mutations (in bold) and other potential pathogenic
variants in distinct population.
|
|
|
| Allele frequencies
in distinct populations |
|---|
|
|
|
|
|
|---|
| Mutation | Nucleotide
change | Amino acid
change | Han | Japanese | African | American | European | All |
|---|
| rs201433665 | c.336+2T>G | p.? | 0.0025 | 0 | 0 | 0 | 0 | <0.001 |
| rs202182550 | c.455G>T | p.? | 0.0025 | 0 | 0 | 0 | 0 | <0.001 |
| rs142859678 | c.796C>T | p.Arg266* | 0.017 | 0.014 | 0 | 0 | 0 | 0.002 |
| rs182539714 | c.-16T>A | p.? | 0.005 | 0.005 | 0 | 0 | 0 | 0.001 |
| rs199666937 | c.140A>G | p.Gln47Arg | 0 | 0 | 0 | 0 | 0.001 | <0.001 |
| rs74653657 | c.181A>G | p.Asn61Asp | 0.012 | 0 | 0 | 0 | 0 | 0.002 |
| rs201239910 | c.388G>A | p.Asp130Asn | 0 | 0 | 0.001 | 0 | 0 | <0.001 |
| rs201821537 | c.219+8C>A | p.? | 0 | 0 | 0 | 0.001 | 0 | <0.001 |
| rs186928560 | c.220-6T>G | p.? | 0 | 0 | 0 | 0 | 0.001 | <0.001 |
| rs139542928 | c.943C>T | p.Arg315Cys | 0 | 0 | 0.001 | 0 | 0 | <0.001 |
| rs201208667 | c.1136G>A | p.Cys379Tyr | 0 | 0 | 0 | 0 | 0.001 | <0.001 |
SERPINB7 is expressed in the epidermis of the whole
body, and belongs to a cluster of clade-B serpins that inhibit
serine proteases and protect cells from exogenous and endogenous
proteolysis (12). Notably,
absence of the critical reactive site loop (P17-P50; amino acid
residues, 331–352) is predicted to be responsible for skin
abnormalities in NPPK. All of the identified mutations, with the
exception of the novel mutation detected in the present cohort, are
expected to truncate the reactive site loop, thus resulting in NPPK
phenotypes. The precise function of SERPINB7 remains poorly
defined. Sakabe et al (13)
speculated that the pathogenesis of NPPK may be associated with the
effects of T cells infiltrating into the skin, thus suggesting a
potential treatment with topical drugs that inhibit T cell
infiltration, such as tacrolimus ointment.
The causal genes of PPK are associated with variable
phenotypes (5). SLURP1
mutations may cause mild MDM without plantar, nail, knee and elbow
involvement, as well as severe complications, including higher
occurrence of malignant melanoma in the hyperkeratotic area
(14). TRPV3 mutations
result in Olmsted syndrome (OMIM #614594) with bilateral mutilating
palmoplantar hyperkeratosis and periorificial keratotic plaques,
along with focal PPK with mild acropodium deformation (15). SERPINB7-associated NPPK has
been reported to exhibit little phenotypic heterogeneity,
corresponding hyperkeratotic lesions with a reddish appearance that
predominantly occur in chronic mechanical stress-exposed areas of
the skin, and a non-progressive disease course (3), all of which were also observed in the
present cohort (Figs. 1–3). These clinical phenomena suggested
that except for the distinct pathogenic mechanisms of corresponding
genes, external stimuli, such as injury and friction, may also be
crucial factors associated with the occurrence of NPPK phenotypes,
which is similar to the findings of our previous report regarding
the progression of MDM (16).
Furthermore, taking into account other potential factors, including
epigenetic alterations, modifier genes and ethnic background,
sometimes diseases with a diffuse PPK appearance are analogous and
hard to differentiate. For instance, the clinical photographs in
previously published studies regarding clinically suspected NPPK
seem to bear close similarity to mild or early stage MDM due to the
yellowish palmoplantar lesions (5,17),
which differ from the usual reddish appearance in pediatric
patients with NPPK (Fig. 2). In
addition, other diseases, such as AREI, Unna-Thost type PPK and
Bothnia type PPK, are difficult to clinically distinguish, which
further underlines the complexity of clinical differential
diagnoses between diseases with palmoplantar hyperkeratosis and
without other associated features, as well as the importance of
molecular analysis.
In conclusion, the present report focused on
patients in Mainland China, with respect to nine definite and three
suspected patients with NPPK. The results revealed three recurrent
and one novel SERPINB7 mutation, thus extending the mutation
spectrum of NPPK. Taking the other seven reported Chinese patients,
that were definitively diagnosed with NPPK by genetic testing, into
account, the present study further demonstrated that NPPK is a
common entity in Mainland China, and c.796C>T is the most
prevalent mutation and exerts a founder effect. At present, there
is no effective drug treatment for the majority of diffuse and
severe forms of PPK, whereas the symptoms of NPPK are relatively
milder and long-time prognosis is favorable. Considering the high
allele frequency of the founder mutation, genetic counseling is
essential for patients with NPPK or carriers of NPPK.
Acknowledgements
The present study was supported by grants from the
Industry Foundation of Ministry of Health of China (grant no.
20120213), the Ph.D. Programs Foundation of Ministry of Education
of China (grant no. 20130073120014), the Natural Science Foundation
of Shanghai Jiaotong University School of Medicine (grant no.
13XJ10023) and a grant from the Foundation of Xinhua Hospital
affiliated to Shanghai Jiaotong University School of Medicine
(grant no. 15YJ15).
References
|
1
|
Kabashima K, Sakabe J, Yamada Y and Tokura
Y: ‘Nagashima-type’ keratosis as a novel entity in the palmoplantar
keratoderma category. Arch Dermatol. 144:375–379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yin J, Xu G, Wang H, Zhao J, Duo L, Cao X,
Tang Z, Lin Z and Yang Y: New and recurrent SERPINB7 mutations in
seven Chinese patients with Nagashima-type palmoplantar keratosis.
J Invest Dermatol. 134:2269–2272. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kubo A, Shiohama A, Sasaki T, Nakabayashi
K, Kawasaki H, Atsugi T, Sato S, Shimizu A, Mikami S, Tanizaki H,
et al: Mutations in SERPINB7, encoding a member of the serine
protease inhibitor superfamily, cause Nagashima-type palmoplantar
keratosis. Am J Hum Genet. 93:945–956. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moosbrugger-Martinz V, Jalili A, Schossig
AS, Jahn-Bassler K, Zschocke J, Schmuth M, Stingl G, Eckl KM,
Hennies HC and Gruber R: Epidermal barrier abnormalities in
exfoliative ichthyosis with a novel homozygous loss-of-function
mutation in CSTA. Brit J Dermatol. 172:1628–1632. 2015. View Article : Google Scholar
|
|
5
|
Gruber R, Hennies HC, Romani N and Schmuth
M: A novel homozygous missense mutation in SLURP1 causing Mal de
Meleda with an atypical phenotype. Arch Dermatol. 147:748–750.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizuno O, Nomura T, Suzuki S, Takeda M,
Ohguchi Y, Fujita Y, Nishie W, Sugiura K, Akiyama M and Shimizu H:
Highly prevalent SERPINB7 founder mutation causes pseudodominant
inheritance pattern in Nagashima-type palmoplantar keratosis. Br J
Dermatol. 171:847–853. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hida T, Okura M, Kamiya T and Yamashita T:
Nagashima-type palmoplantar keratosis caused by compound
heterozygous mutations in SERPINB7. Eur J Dermatol. 25:202–203.
2015.PubMed/NCBI
|
|
8
|
Mokni M, Charfeddine C, Ben Mously R,
Baccouche D, Kaabi B, Ben Osman A, Dellagi K and Abdelhak S:
Heterozygous manifestations in female carriers of Mal de Meleda.
Clin Genet. 65:244–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eckl KM, Stevens HP, Lestringant GG,
Westenberger-Treumann M, Traupe H, Hinz B, Frossard PM, Stadler R,
Leigh IM, Nürnberg P, et al: Mal de Meleda (MDM) caused by
mutations in the gene for SLURP-1 in patients from Germany, Turkey,
Palestine, and the United Arab Emirates. Hum Genet. 112:50–56.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bchetnia M, Laroussi N, Youssef M,
Charfeddine C, Ben Brick AS, Boubaker MS, Mokni M, Abdelhak S, Zili
J and Benmously R: Particular Mal de Meleda phenotypes in Tunisia
and mutations founder effect in the Mediterranean region. Biomed
Res Int. 2013:2068032013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kubo A: Nagashima-type palmoplantar
keratosis: A common Asian type caused by SERPINB7 protease
inhibitor deficiency. J Invest Dermatol. 134:2076–2079. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Silverman GA, Whisstock JC, Askew DJ, Pak
SC, Luke CJ, Cataltepe S, Irving JA and Bird PI: Human clade B
serpins (ov-serpins) belong to a cohort of evolutionarily dispersed
intracellular proteinase inhibitor clades that protect cells from
promiscuous proteolysis. Cell Mol Life Sci. 61:301–325. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakabe JI, Kabashima K, Sugita K and
Tokura Y: Possible involvement of T lymphocytes in the pathogenesis
of Nagashima-type keratosis palmoplantaris. Clin Exp Dermatol.
34:e282–e284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sartore L, Bordignon M, Bassetto F, Voltan
A, Tomat V and Alaibac M: Melanoma in skin affected with
keratoderma palmoplantaris hereditaria (Mal de Meleda): Treatment
with excision and grafting. J Am Acad Dermatol. 61:161–163. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
He Y, Zeng K, Zhang X, Chen Q, Wu J, Li H,
Zhou Y, Glusman G, Roach J, Etheridge A, et al: A gain-of-function
mutation in TRPV3 causes focal palmoplantar keratoderma in a
Chinese family. J Invest Dermatol. 135:907–909. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Cheng R, Ni C, Liang J, Li M and
Yao Z: First Mal de Meleda report in Chinese Mainland: Two families
with a recurrent homozygous missense mutation in SLURP-1. J Eur
Acad Dermatol Venereol. 30:871–873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nakamizo S, Katoh N, Miyachi Y and
Kabashima K: Atypical nail dystrophy in a possible case of
Nagashima-type palmoplantar keratosis. J Dermatol. 39:470–471.
2012. View Article : Google Scholar : PubMed/NCBI
|