Introduction
Neurofibromatosis type 1 (NF1; OMIM 162200) is one
of the most common hereditary disorders. It is predominantly
characterized by multiple café-au-lait macules (CALM), skin-fold
freckling, Lisch nodules and neurofibromas. However, as the
condition exhibits age-dependent characteristics and there are a
number of other overlapping syndromes and similar diseases, it is
usually difficult to make an early clinical diagnosis. Currently,
although there are numerous comprehensive detection methods
available for molecular diagnosis of NF1, the highest sensitivity
of any of these methods is ~95% (1,2). In
addition to the limitations of detection by these approaches, cases
of mosaic NF1 have been reported and there may exist other known or
undefined genetic conditions with similar phenotypes. KIT ligand
(KITLG) and its receptor KIT proto-oncogene receptor tyrosine
kinase (KIT) activate the Ras/mitogen-activated protein kinase
(MAPK) signaling pathway and they are critical in the control of
physiological and pathological cutaneous pigmentation, including
CALM (3). Known disorders with
CALM comprise: i) A group of genetic syndromes resulting from
germline mutations in genes that encode components or regulators of
the Ras/MAPK signaling pathway, designated RASopathies, which
consists of Noonan syndrome (NS; OMIM 163950), Legius syndrome
[formerly termed NF1-like syndrome (NFLS); OMIM 611431], LEOPARD
syndrome (LS; OMIM 151100), Costello syndrome (CS; OMIM 218040) and
cardiofaciocutaneous syndrome (CFC; OMIM 115150), ii) disorders
involving the KITLG/KIT signaling pathway, including piebaldism
(OMIM 172800) and familial progressive hyperpigmentation (FPH; OMIM
145250). Although there are unique phenotypic characteristics for
these conditions, certain syndromes remain highly overlapping and
the differential diagnosis between them and NF1 is complex.
Our group recently conducted a large molecular
research investigation into NF1 in a Chinese population (4–6).
Patients without NF1 mutations who exhibited CALM were
observed, and in a further study, other syndromes were
demonstrated, including an atypical LS patient with a PTPN11
mutation, a piebaldism patient with a KIT mutation and a
familial progressive hyper- and hypopigmentation (FPHH) patient
with a KITLG mutation.
The present review discusses these KITLG/c-Kit- and
Ras/MAPK signaling pathway-associated ‘NF1-like’ inherited
diseases, and proposes a molecular screening strategy to aid the
determination of a definitive diagnosis.
The RASopathies with CALM
NFLS
NFLS presents predominantly with CALM,
intertriginous freckling and certain less common manifestations
(~20%), including neurocognitive impairment, developmental delay
and macrocephaly, usually without neurofibromas, Lisch nodules,
optic pathway glioma and other tumors (7,8).
More than 60% of the patients have a family history of the
condition (9). These features meet
three of the National Institutes of Health (NIH) diagnostic
criteria for NF1 (10), thus, it
is not reliable to distinguish NF1 from NFLS relying solely on
clinical examination. Germline loss-of-function sprouty related,
EVH1 domain containing 1 (SPRED1) mutations are responsible
for this syndrome, which is most similar to NF1 in early childhood
(11). It is estimated that ~1–4%
of individuals with multiple CALM harbor heterozygous SPRED1
mutations (7,9). In subjects with familial CALM, with
or without freckling and no other NF1 features, 73% and 19% carry
pathogenic NF1 mutations or SPRED1 mutations,
respectively (7), which further
indicates that genetic testing is useful in diagnosis in these
cases (92%). SPRED1 is involved in regulation of the MAPK signaling
pathway, previous studies have demonstrated a loss of
heterozygosity in pediatric acute myeloblastic leukemia, acting as
a tumor suppressor (12,13), this also indicates that this
syndrome increases the risk of developing specific hematological
malignancies, as well as other rare conditions, including kidney
and lung cancer (8).
LS
LS is an autosomal dominant RASopathy, predominantly
caused by mutations in protein tyrosine phosphatase, non-receptor
type 11 (PTPN11; 85%), in addition to less common genes,
Raf-1 proto-oncogene, serine/threonine kinase (RAF1) and B-raf
proto-oncogene, serine/threonine kinase (BRAF) (10%).
Individuals present with characteristic multiple lentigines, and
relatively common features of RASopathies, including facial
dysmorphia, myocardial and valvular abnormalities, and hearing
loss. It is hard to clinically distinguish LS from other
RASopathies, such as NF1 and NS, while molecular diagnosis is
relatively reliable.
Twelve different missense PTPN11 mutations
(Tyr279Cys/Ser, Ala461Thr/Ser, Gly464Ala, Thr468Met/Pro,
Arg498Leu/Trp, Gln506Pro and Gln510Glu/Pro) (14–18)
were reported to result in LS. Two of these mutations (Tyr279Cys
and Thr468Met) account for ~65% of the cases. Notably, these
mutations cluster in the catalytic protein tyrosine phosphatase
domain (amino acid residues, 221–524) (19), as in the allelic disorder, NS, the
majority of missense mutations, small deletions and indels
(20,21) are associated with the N-SH2 domain
(amino acid residues, 3–104) (14). Contrary to the gain-of-function
changes resulting in excessive PTPN11 activity in NS (12), LS mutants are catalytically
defective and exert a dominant negative effect (22), suggesting that mutation type and
region are important in the underlying pathogenic mechanisms and
differential diagnoses of NS and LS. A recent study has suggested
that LS-associated mutations may increase melanin synthesis in
melanocytes via the activation of Akt/mammalian target of rapamycin
signaling, thus, resulting in a phenotype with multiple lentigines
(23).
Furthermore, a recent study identified a novel
heterozygous MAPK kinase 1 (MAP2K1) mutation in LS,
Glu102Gly (24), this is notable
as mutations in this gene are usually associated with CFC. LS
shares numerous phenotypic traits with CFC. Including this case, at
present, germline mutations in MAP2K1 and BRAF genes
are associated with CFC and LS. CALM and multiple nevi or
lentigines are rare or absent in CFC patients with Kirsten rat
sarcoma viral oncogene homolog (KRAS) mutations (25), further demonstrating the
complicated genetic heterogeneity and prominent overlapping feature
of RASopathies.
CALM precede or are associated with lentigines
in~10% of NS cases (26), and in
up to 75% of LS cases (27),
furthermore, in LS cases, the number of CALM can fulfills NIH
criteria for diagnosis with NF1 (28). Lentigines usually appear during
childhood as black-brown macules, predominantly on the face, neck
and upper part of the trunk, and gradually increase in number and
darken in color with age. Although LS patients have unclear
disposition to malignancies, certain studies have reported an
association with hematologic malignancies (29) and medulloblastoma (30), which should be noted.
NS-CFC-CS spectrum
NS shares numerous congenital anomalies with LS,
excluding multiple lentigines. CFC and CS also have various
clinical similarities and few differences compared with NS
(25). Previously, clinical
discrimination between these three syndromes was predominantly
based on respective characteristic features, including
hyperkeratotic skin, ichthyosis and keratosis pilaris in CFC
patients; and soft and loose skin, deep palmar/plantar creases,
nasal papillomas and an increased risk of developing malignancies
in those with CS (31,32). However, as the reported cases of
RASopathies increase, these distinct features have also become
highly overlapped.
Previous studies have demonstrated that one or two
CALM are observed in 9–31% of individuals in NS-CFC-CS spectrum
disorders, this is markedly higher than the overall prevalence of
2.5% in neonates (33), while
multiple CALM and intertriginous freckling were rare or absent
(25,26,34).
In combination with the less common dysmorphic craniofacial
features in NF1, the discrimination between these conditions and
NF1 is relatively easier.
KRAS mutations associated with the NS-CFC-CS
spectrum predominantly confer mild gain-of-function effect
(35). The present review suggests
that KRAS mutations associated with the NS-CFC-CS spectrum
belong to an identical entity relatively close to NS (see Table I, which provides a proper
hypothesis for a new classification of these RASopathies and
corresponding causal genes) as: i) CFC, NS and CS exhibit numerous
clinical similarities and few differences (25,36,37);
ii) KRAS mutations have been reported in <2% of the NS
and CFC cases (37), as well as in
only a handful of CS patients (35,38),
and all three of the independent conditions demonstrated their
major pathogenic form, PTPN11, BRAF and HRAS,
respectively (presented in Table
I); iii) mutations Asp153Val, Thr58Ile and other missense
mutations in the same amino acid residue in KRAS were
reported to result in NS and CFC (25,39,40);
iv) less prominent ectodermal phenotypes were observed in CFC and
CS with KRAS mutations than those with BRAF mutations
(25,34,39,41);
v) multiple nevi or lentigines were rare or absent in CFC patients
with KRAS mutations compared with those with mutations in
MAP2K1 and BRAF (25).
 | Table I.‘NF1-like’ genetic disorders with
CALM involved in KITLG/c-Kit and Ras/MAPK signaling pathways. |
Table I.
‘NF1-like’ genetic disorders with
CALM involved in KITLG/c-Kit and Ras/MAPK signaling pathways.
| Genetic
disorder | Known causal genes
(proportion) | Disease
identity | Gene identity | Characteristic
features |
|---|
| NS | PTPN11 (50%) SOS1
(10–15%) RAF1 (5%) RIT1 (5%) KRAS (<2%) BRAF (rare) NRAS (rare)
SHOC2 (rare) CBL (rare) | NS | PTPN11 SOS1 RAF1
RIT1 NRAS SHOC2 CBL | CALM (10%).
Dysmorphic craniofacial features, cardiac defect (pulmonary valve
stenosis, hypertrophic cardiomyopathy), musculoskeletal
abnormalities, mental retardation, cryptorchidism, hematologic
malignancies |
|
|
|
KRAS-associated NS-CFC-CS
spectrum | KRAS | Less prominent
ectodermal phenotypes; multiple nevi or lentigines were rare or
absent |
| CFC | BRAF (50–70%)
MAP2K1/2 (25%) KRAS (<2%) | CFC | BRAF MAP2K1/2 | CALM (9–31%).
Similar to NS. Ectodermal abnormalities such as multiple nevi,
keratosis pilaris, ulerythema ophryogenes and brittle, sparse,
curly hair. Potential cancer risk |
| CS | HRAS (>90%) BRAF
(rare) KRAS (rare) | CS | HRAS | CALM (rare).
Similar to NS. Ectodermal abnormalities like soft skin, deep
palmar/plantar creases, papillomas and curly hair. Severe failure
to thrive. Significant cancer risk (17%) |
| LS | PTPN11 (85%) RAF1
(rare) BRAF (rare) MAP2K1 (1 case) |
|
| CALM (70–80%).
Similar to NS, but with multiple lentigines mostly on face, neck
and upper part of the trunk. Unclear cancer risk |
| NFLS | SPRED1 |
|
| Multiple CALM
(nearly 100%), intertriginous freckling. Potential risk of
pediatric AML |
| Piebaldism | KIT |
|
| Depigmented patches
of skin and hair |
| FPH and FPHH | KITLG | FPHH | KITLG | Diffuse, partly
blotchy hyperpigmented lesions intermingled with scattered
hypopigmentations, lentigines and CALM |
In the majority of instances, BRAF mutations
resulting in NS have not been observed in CFC, suggesting the
associated phenotypes may be allele specific (42), however, a number of BRAF
mutations, including Ala246Pro and Gln257Arg, have been
demonstrated in the two conditions (25).
Previous studies have also demonstrated the
evolution of the clinical phenotype in a CFC patient, and the
marked resemblance between CFC and NS, consistent with the
suggestion that NS and CFC are variable manifestations of the same
entity (36,43).
Taking updated molecular findings, reviews of
complex genetic heterogeneity and the highly overlapping features
of these disorders into consideration, the present review
hypothesizes these three disorders may be not distinct and separate
conditions, but a continuous spectrum consisting of a certain gene
or group of gene-related subtypes with certain degrees of
phenotypic variability, particularly KRAS-associated
NS-CFC-CS spectrum (as presented in Table I), or multiple alternative
underlying mechanisms are involved in the functional dysregulation
of the Ras/MAPK signaling pathway.
Allelic syndromes of NF1:
Neurofibromatosis-Noonan syndrome and Watson syndrome
The disorder designated neurofibromatosis-Noonan
syndrome (NFNS; OMIM 601321) is a variant of NF1 rather than NS,
predominantly due to mutations in the NF1 gene (44). It may fulfill the criteria for NF1
with CALM and skin-fold freckling, but also has overlapping
features with NS, including ‘Noonan’ face, short stature,
congenital heart defects and a predisposition to malignancy.
Watson syndrome (WS; OMIM 193520) is characterized
by pulmonic stenosis, CALM and intellectual impairment (45), furthermore, Lisch nodules are
observed in the majority of affected subjects, and neurofibromas in
~1/3 (46). An 80-kb deletion and
an in-frame tandem duplication of 42 bases at the NF1 locus
have been reported in patients with WS (47,48).
These findings broaden the noteworthy NF1-associated
phenotype spectrum and are consistent with NFNS and WS as allelic
disorders or subtypes of NF1. An alternative explanation is that
they are the result of an additive effect of mutations in
NF1 and other relevant genes, including PTPN11 or
unknown modifying loci (49,50).
KITLG/KIT signaling pathway-associated
genetic disorders with CALM
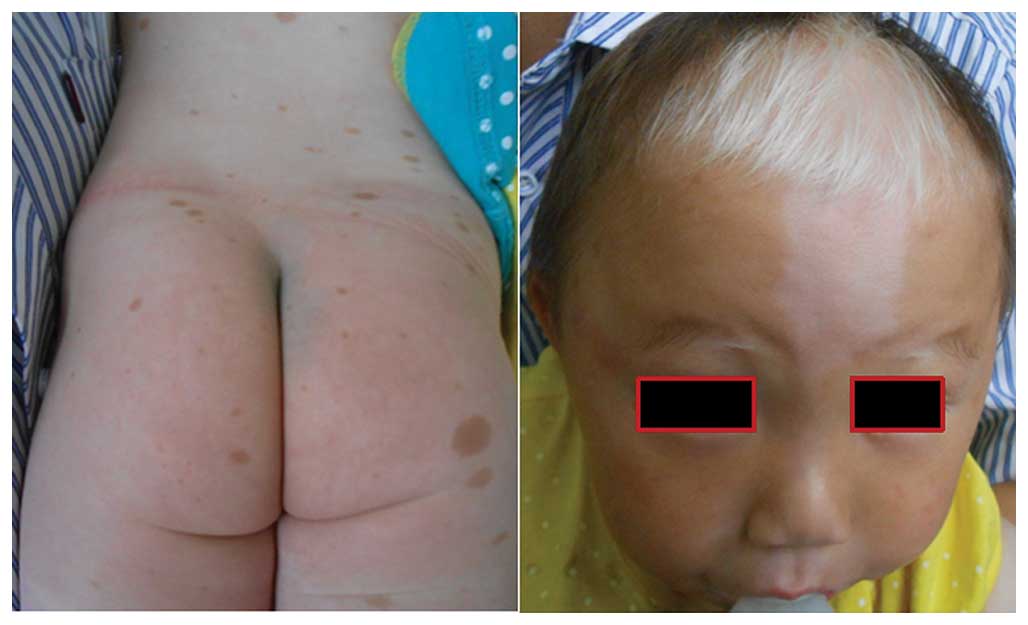
Piebaldism
Piebaldism is a rare autosomal dominant disorder
caused by KIT mutations. Characteristic features are
depigmented patches of skin and hair (as presented in Fig. 1). Of the three reported piebaldism
cases with multiple CALM and intertriginous freckling, all the
mutations were located in the tyrosine kinase (TK) domain
(Gly610Asp, Gliu640Asp and Arg791Gly) (51–53).
It has been demonstrated that inadequate phosphorylation of the
KIT-binding domain in SPRED1 due to a defective KIT TK would result
in loss of inhibition of the Ras⁄MAPK signaling pathway, leading to
a phenotype similar to NFLS (53),
while gain-of function mutations in KITLG have been reported to
result in FPHH (54), indicating
KIT and KITLG are important modulators of skin pigmentation.
Phenotypic severity depends on the type and site of
the mutation (55,56). Mutations in the TK region (TK1,
582–684 and TK2, 762–973) exert a dominant-negative effect, usually
resulting in a severe phenotype, whereas mild cases are frequently
due to mutations in the extracellular region.
Patients with piebaldism may develop CALM (as
presented in Fig. 1), NF1 may be
associated with piebaldism, and these two distinct conditions may
co-exist in one patient (57,58),
which highlights the necessity of molecular diagnosis.
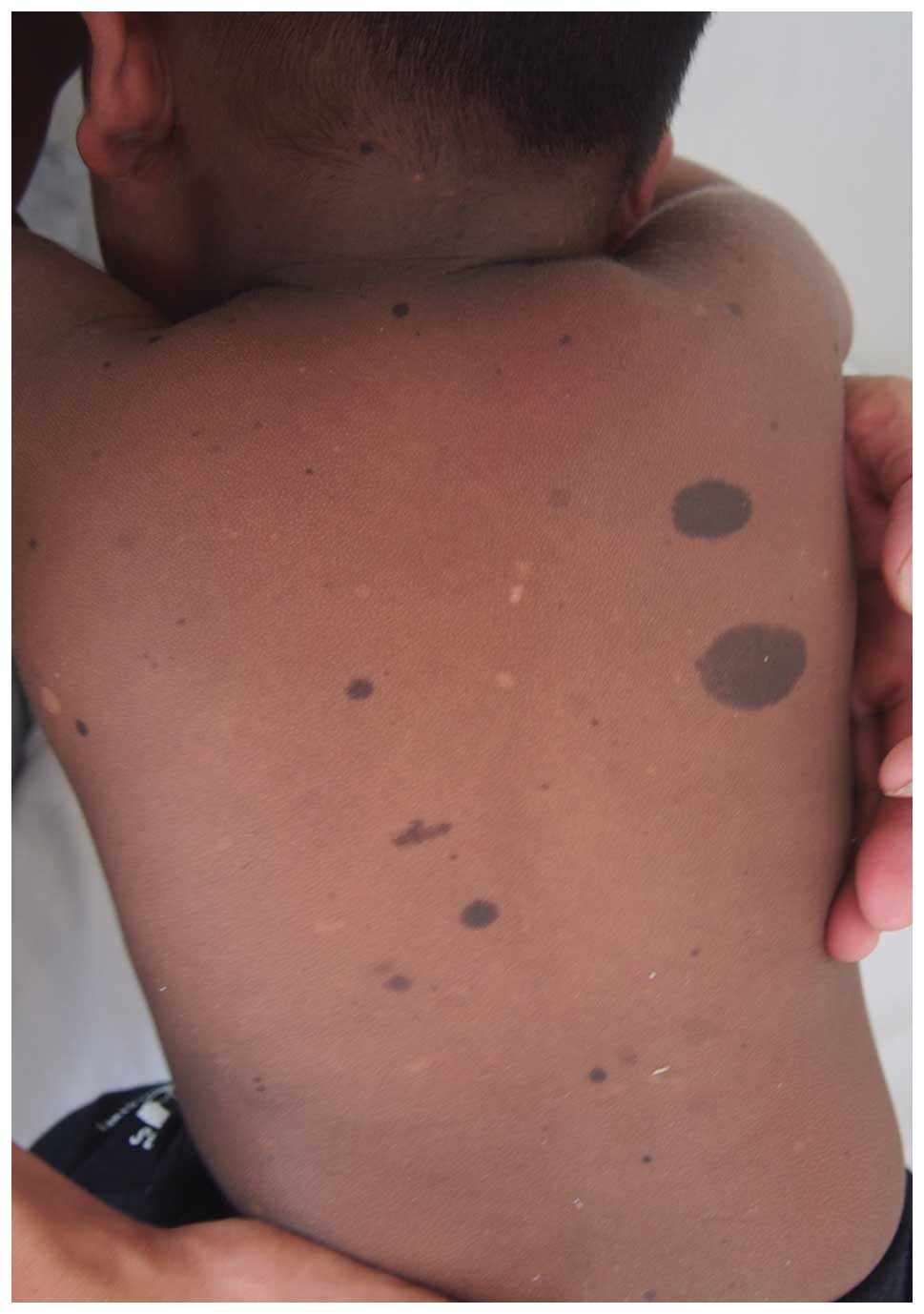
FPHH and FPH
FPHH is notable for progressive, diffuse, partly
blotchy hyperpigmented lesions intermingled with scattered
hypopigmented spots, lentigines and CALM (as presented in Fig. 2) (6). It is the result of a mutation in
KITLG, encoding KITLG involved in the Ras⁄MAPK signaling
pathway (54). Clinical signs are
somewhat different from its allelic disorder FPH (59), in which no hypopigmentation is
present. Notably, the mutation p.Asn36Ser results in FPH and FPHH,
with the FPH patient image in a previous study by Wang et al
(59) also demonstrating small
suspicious hypopigmented lesions (54). This suggests these two disorders
may resemble another pigmentary genetic disorder termed
Dowling-Degos disease (DDD; OMIM 179850, 615327 and 615696)
(60–63), which may also be the same condition
with a degree of phenotypic variability, for example, in the
distribution of hyperpigmented and hypopigmented lesions.
Conclusion
The RASopathies have complex genetic heterogeneity
and marked overlapping features, however, a relatively correct
diagnosis is essential for genetic counseling regarding prognosis
(as the diagnosis of milder phenotypes, including NFLS or mosaic
NF1, may relieve the psychological burden on serious age-dependent
complications); monitoring of potential risks, including cancer and
cardiac events; and prevention using prenatal diagnosis.
Thus, the current review proposes a screening
strategy in which: i) NF1 testing has a priority for
patients that only exhibit CALM or fulfill the NIH diagnostic
criteria for NF1; ii) SPRED1 and NF1 should be tested
in those with skin-fold freckling and a family history; iii) in
NF1-negative pediatric patients with CALM and a dispersed
pattern of facial and cervical freckles, PTPN11 should be
first considered for molecular analysis, as it accounts for the
majority of LS and NS cases; iv) genetic testing of KITLG
and KIT is used and relatively effective in those with
diffuse hyperpigmented lesions intermingled with scattered
depigmentation, depigmented patches of skin and hair, respectively
(as presented in Fig. 3).
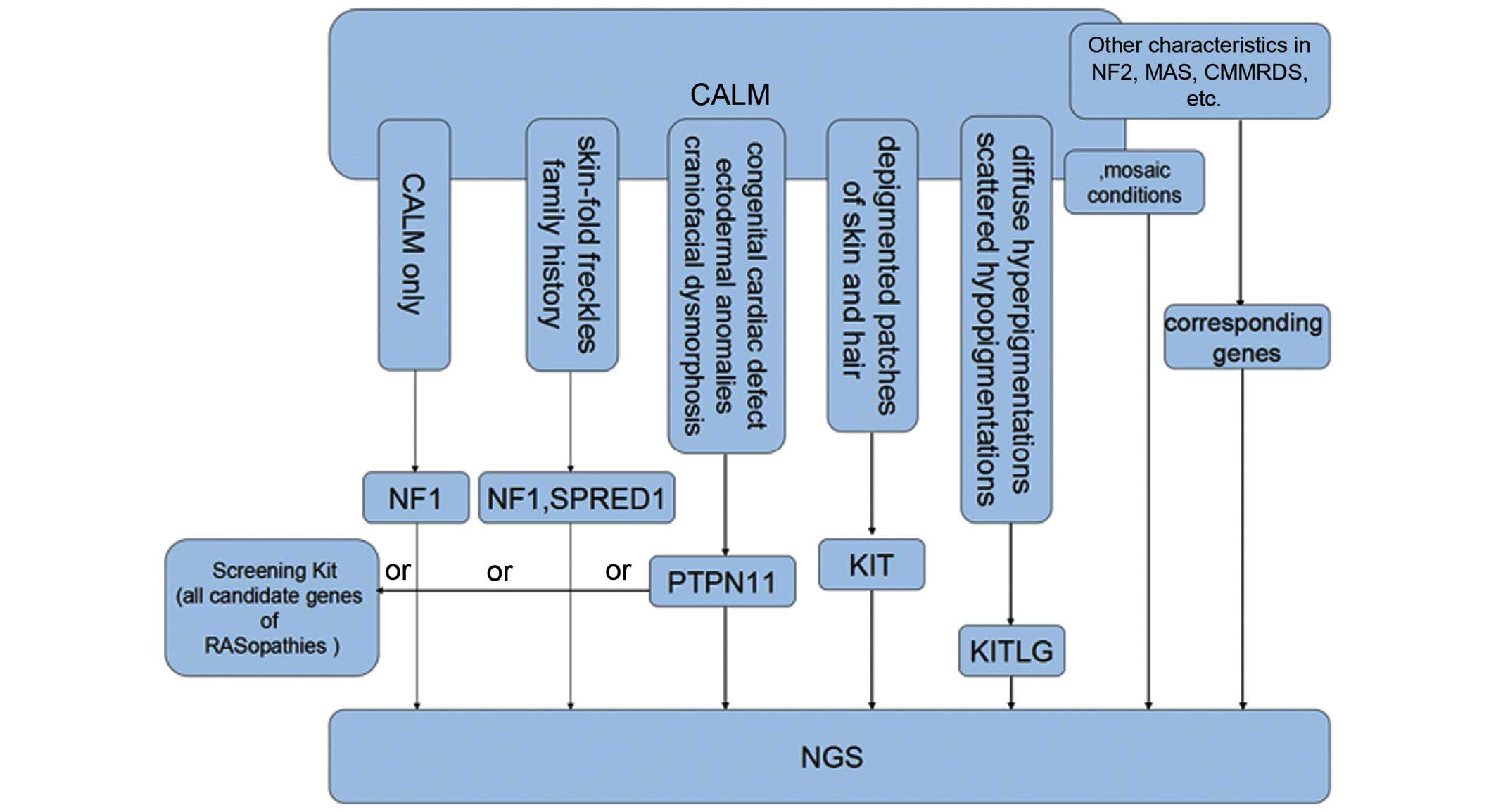 | Figure 3.Screening strategies for inherited
disorders with CALM that clinically resemble NF1, particularly
those involved in KITLG/KIT and Ras/mitogen-activated protein
kinase signaling pathways. CALM, café-au-lait macules; NF1,
neurofibromatosis type 1; KIT, KIT proto-oncogene receptor tyrosine
kinase; KITLG, KIT ligand; SPRED1, sprouty related, EVH1 domain
containing 1; PTPN11, protein tyrosine phosphatase, non-receptor
type 11; NF2, neurofibromatosis type 2; MAS. McCune-Albright
syndrome; CMMRDS, constitutional mismatch repair deficiency; NGS,
next generation sequencing. |
As for other RASopathies, BRAF and
HRAS analysis is suitable for relatively typical CFC and CS
presentations, respectively. However, when considering the highly
overlapping phenotypes and involvement of numerous genes, a
custom-designed screening kit including all the candidate genes for
the various RASopathies is a thorough alternative choice.
Furthermore, few patients with CALM fall outside the
above mentioned disorders and their condition may be the result of
mutations in other undetected genes associated with the Ras/MAPK
signaling pathway, for instance, recently identified causal genes
Cbl proto-oncogene E3 ubiquitin protein ligase and Ras-like without
CAAX 1 (64,65). Considering the detection limit of
general sequencing methods, especially for those atypical or
unreported phenotypes, next generation sequencing (such as whole
exome sequencing and whole genome sequencing) still serves as a
cost-effective approach for molecular diagnosis of the above
disorders with CALS, as well as other possible genetic diseases,
including neurofibromatosis type 2 (OMIM 101000) with vestibular
schwannomas; McCune-Albright syndrome (OMIM 174800) with segmental
CALM, polyostotic fibrous dysplasia and precocious puberty;
constitutional mismatch repair deficiency syndrome (OMIM 276300)
with childhood cancer predisposition (66); and various mosaic conditions
associated with CALM (as presented in Fig. 3).
Acknowledgements
The current review was supported by a grant from the
Ph.D. Programs Foundation of Ministry of Education of China (grant
no. 20130073120014), a grant from the Natural Science Foundation of
Shanghai Jiaotong University School of Medicine (grant no.
13XJ10023) and a grant from the Foundation of Xinhua Hospital
Affiliated to Shanghai Jiaotong University School of Medicine
(grant no. 15YJ15).
References
|
1
|
Messiaen LM, Callens T, Mortier G, Beysen
D, Vandenbroucke I, Van Roy N, Speleman F and Paepe AD: Exhaustive
mutation analysis of the NF1 gene allows identification of 95% of
mutations and reveals a high frequency of unusual splicing defects.
Hum Mutat. 15:541–555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Valero MC, Martin Y, Hernández-Imaz E,
Marina Hernández A, Meleán G, Valero AM, Javier Rodríguez-Álvarez
F, Tellería D and Hernández-Chico C: A highly sensitive genetic
protocol to detect NF1 mutations. J Mol Diagn. 13:113–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Picardo M and Cardinali G: The genetic
determination of skin pigmentation: KITLG and the KITLG/c-Kit
pathway as key players in the onset of human familial pigmentary
diseases. J Invest Dermatol. 131:1182–1185. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang J, Tong H, Fu X, Zhang Y, Liu J,
Cheng R, Liang J, Peng J, Sun Z, Liu H, et al: Molecular
characterization of NF1 and neurofibromatosis type 1
genotype-phenotype correlations in a Chinese population. Sci Rep.
5:112912015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang J, Cheng R, Liang J, Ni C, Li M and
Yao Z: Lentiginous phenotypes caused by diverse pathogenic genes
(SASH1 and PTPN11): Clinical and molecular discrimination. Clin
Genet. Feb 3–2016.Epub ahead of print. View Article : Google Scholar
|
|
6
|
Zhang J, Cheng R, Liang J, Ni C, Li M and
Yao Z: Report of a sporadic familial progressive hyper- and
hypopigmentation child caused by a novel KITLG mutation. Br J
Dermatol. Apr 23–2016.Epub ahead of print. View Article : Google Scholar
|
|
7
|
Messiaen L, Yao S, Brems H, Callens T,
Sathienkijkanchai A, Denayer E, Spencer E, Arn P,
Babovic-Vuksanovic D, Bay C, et al: Clinical and mutational
spectrum of neurofibromatosis type 1-like syndrome. JAMA.
302:2111–2118. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brems H, Pasmant E, Van Minkelen R, Wimmer
K, Upadhyaya M, Legius E and Messiaen L: Review and update of
SPRED1 mutations causing Legius syndrome. Hum Mutat. 33:1538–1546.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Brems H and Legius E: Legius syndrome, an
update. Molecular pathology of mutations in SPRED1. Keio J Med.
62:107–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
[No authors lsited]: Neurofibromatosis.
Conference statement. National Institutes of Health Consensus
Development Conference, . Arch Neurol. 45:575–578. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brems H, Chmara M, Sahbatou M, Denayer E,
Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP,
et al: Germline loss-of-function mutations in SPRED1 cause a
neurofibromatosis 1-like phenotype. Nat Genet. 39:1120–1126. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pasmant E, Gilbert-Dussardier B, Petit A,
de Laval B, Luscan A, Gruber A, Lapillonne H, Deswarte C, Goussard
P, Laurendeau I, et al: SPRED1, a RAS MAPK pathway inhibitor that
causes Legius syndrome, is a tumour suppressor downregulated in
paediatric acute myeloblastic leukaemia. Oncogene. 34:631–638.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pasmant E, Ballerini P, Lapillonne H,
Perot C, Vidaud D, Leverger G and Landman-Parker J: SPRED1 disorder
and predisposition to leukemia in children. Blood. 114:11312009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tartaglia M, Mehler EL, Goldberg R,
Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A,
Jeffery S, et al: Mutations in PTPN11, encoding the protein
tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet.
29:465–468. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Conti E, Dottorini T, Sarkozy A, Tiller
GE, Esposito G, Pizzuti A and Dallapiccola B: A novel PTPN11
mutation in LEOPARD syndrome. Hum Mutat. 21:6542003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martínez-Quintana E and Rodríguez-González
F: LEOPARD Syndrome: Clinical features and gene mutations. Mol
Syndromol. 3:145–157. 2012.PubMed/NCBI
|
|
17
|
Wang Y, Chen C and Wang DW: Leopard
syndrome caused by heterozygous missense mutation of Tyr 279 Cys in
the PTPN11 gene in a sporadic case of Chinese Han. Int J Cardiol.
174:e101–e104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Osawa R, Akiyama M, Yamanaka Y, Ujiie H,
Nemoto-Hasebe I, Takeda A, Yanagi T and Shimizu H: A novel PTPN11
missense mutation in a patient with LEOPARD syndrome. Brit J
Dermatol. 161:1202–1204. 2009. View Article : Google Scholar
|
|
19
|
Digilio MC, Conti E, Sarkozy A, Mingarelli
R, Dottorini T, Marino B, Pizzuti A and Dallapiccola B: Grouping of
multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11
gene. Am J Hum Genet. 71:389–394. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoshida R, Hasegawa T, Hasegawa Y, Nagai
T, Kinoshita E, Tanaka Y, Kanegane H, Ohyama K, Onishi T, Hanew K,
et al: Protein-tyrosine phosphatase, nonreceptor type 11 mutation
analysis and clinical assessment in 45 patients with Noonan
syndrome. J Clin Endocrinol Metab. 89:3359–3364. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tartaglia M, Martinelli S, Stella L,
Bocchinfuso G, Flex E, Cordeddu V, Zampino G, Burgt Iv, Palleschi
A, Petrucci TC, et al: Diversity and functional consequences of
germline and somatic PTPN11 mutations in human disease. Am J Hum
Genet. 78:279–290. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kontaridis MI, Swanson KD, David FS,
Barford D and Neel BG: PTPN11 (Shp2) mutations in LEOPARD syndrome
have dominant negative, not activating, effects. J Biol Chem.
281:6785–6792. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Motegi S, Yokoyama Y, Ogino S, Yamada K,
Uchiyama A, Perera B, Takeuchi Y, Ohnishi H and Ishikawa O:
Pathogenesis of multiple lentigines in LEOPARD syndrome with PTPN11
gene mutation. Acta Derm Venereol. 95:978–984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nishi E, Mizuno S, Nanjo Y, Niihori T,
Fukushima Y, Matsubara Y, Aoki Y and Kosho T: A novel heterozygous
MAP2K1 mutation in a patient with Noonan syndrome with multiple
lentigines. Am J Med Genet A 167A. 407–411. 2015. View Article : Google Scholar
|
|
25
|
Nava C, Hanna N, Michot C, Pereira S,
Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler B, Lacombe D,
et al: Cardio-facio-cutaneous and Noonan syndromes due to mutations
in the RAS/MAPK signalling pathway: Genotype-phenotype
relationships and overlap with Costello syndrome. J Med Genet.
44:763–771. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Allanson JE: Noonan syndrome. J Med Genet.
24:9–13. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Digilio MC, Sarkozy A, de Zorzi A, Pacileo
G, Limongelli G, Mingarelli R, Calabrò R, Marino B and Dallapiccola
B: LEOPARD syndrome: Clinical diagnosis in the first year of life.
Am J Med Genet A. 140:740–746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carcavilla A, Pinto I, Muñoz-Pacheco R,
Barrio R, Martin-Frias M and Ezquieta B: LEOPARD syndrome (PTPN11,
T468 M) in three boys fulfilling neurofibromatosis type 1 clinical
criteria. Eur J Pediatr. 170:1069–1074. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tartaglia M, Niemeyer CM, Fragale A, Song
X, Buechner J, Jung A, Hählen K, Hasle H, Licht JD and Gelb BD:
Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia,
myelodysplastic syndromes and acute myeloid leukemia. Nat Genet.
34:148–150. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rankin J, Short J, Turnpenny P, Castle B
and Hanemann CO: Medulloblastoma in a patient with the PTPN11
p.Thr468Met mutation. Am J Med Genet A 161A. 2027–2029. 2013.
View Article : Google Scholar
|
|
31
|
Hennekam RC: Costello syndrome: An
overview. Am J Med Genet Part C Semin Med Genet 117C. 42–48. 2003.
View Article : Google Scholar
|
|
32
|
Bryan ZT, Missall TA, Stieren S, Siegfried
E and Burkemper NM: Clinicopathologic evaluation of
cardiofaciocutaneous syndrome: Overcoming the challenges of
diagnosing a rare genodermatosis. Pediatr Dermatol. 32:e23–e28.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Alper J, Holmes LB and Mihm MC Jr:
Birthmarks with serious medical significance: Nevocellular nevi,
sebaceous nevi, and multiple café au lait spots. J Pediatr.
95:696–700. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Siegel DH, McKenzie J, Frieden IJ and
Rauen KA: Dermatological findings in 61 mutation-positive
individuals with cardiofaciocutaneous syndrome. Brit J Dermatol.
164:521–529. 2011.
|
|
35
|
Zenker M, Lehmann K, Schulz AL, Barth H,
Hansmann D, Koenig R, Korinthenberg R, Kreiss-Nachtsheim M,
Meinecke P, Morlot S, et al: Expansion of the genotypic and
phenotypic spectrum in patients with KRAS germline mutations. J Med
Genet. 44:131–135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fryer AE, Holt PJ and Hughes HE: The
cardio-facio-cutaneous (CFC) syndrome and Noonan syndrome: Are they
the same? Am J Med Genet. 38:548–551. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tidyman WE and Rauen KA: Noonan, Costello
and cardio-facio-cutaneous syndromes: Dysregulation of the Ras-MAPK
pathway. Expert Rev Mol Med. 10:e372008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bertola DR, Pereira AC, Brasil AS, Albano
LM, Kim CA and Krieger JE: Further evidence of genetic
heterogeneity in Costello syndrome: Involvement of the KRAS gene. J
Hum Genet. 52:521–526. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Niihori T, Aoki Y, Narumi Y, Neri G, Cavé
H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G,
Wieczorek D, et al: Germline KRAS and BRAF mutations in
cardio-facio-cutaneous syndrome. Nat Genet. 38:294–296. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schubbert S, Zenker M, Rowe SL, Böll S,
Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner
LE, et al: Germline KRAS mutations cause Noonan syndrome. Nat
Genet. 38:331–336. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abe Y, Aoki Y, Kuriyama S, Kawame H,
Okamoto N, Kurosawa K, Ohashi H, Mizuno S, Ogata T, Kure S, et al:
Prevalence and clinical features of Costello syndrome and
cardio-facio-cutaneous syndrome in Japan: Findings from a
nationwide epidemiological survey. Am J Med Genet A 158A.
1083–1094. 2012. View Article : Google Scholar
|
|
42
|
Sarkozy A, Carta C, Moretti S, Zampino G,
Digilio MC, Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V,
Lepri F, et al: Germline BRAF mutations in Noonan, LEOPARD, and
cardiofaciocutaneous syndromes: Molecular diversity and associated
phenotypic spectrum. Hum Mutat. 30:695–702. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Neri G and Zollino M: More on the
Noonan-CFC controversy. Am J Med Genet. 65:1001996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
De Luca A, Bottillo I, Sarkozy A, Carta C,
Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A,
et al: NF1 gene mutations represent the major molecular event
underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet.
77:1092–1101. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Watson GH: Pulmonary stenosis,
café-au-lait spots, and dull intelligence. Arch Dis Child.
42:303–307. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Allanson JE, Upadhyaya M, Watson GH,
Partington M, MacKenzie A, Lahey D, MacLeod H, Sarfarazi M,
Broadhead W, Harper PS, et al: Watson syndrome: Is it a subtype of
type 1 neurofibromatosis? J Med Genet. 28:752–756. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Upadhyaya M, Shen M, Cherryson A, Farnham
J, Maynard J, Huson SM and Harper PS: Analysis of mutations at the
neurofibromatosis 1 (NF1) locus. Hum Mol Genet. 1:735–740. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tassabehji M, Strachan T, Sharland M,
Colley A, Donnai D, Harris R and Thakker N: Tandem duplication
within a neurofibromatosis type 1 (NF1) gene exon in a family with
features of Watson syndrome and Noonan syndrome. Am J Hum Genet.
53:90–95. 1993.PubMed/NCBI
|
|
49
|
Thiel C, Wilken M, Zenker M, Sticht H,
Fahsold R, Gusek-Schneider GC and Rauch A: Independent NF1 and
PTPN11 mutations in a family with neurofibromatosis-Noonan
syndrome. Am J Med Genet A 149A. 1263–1267. 2009. View Article : Google Scholar
|
|
50
|
Nyström AM, Ekvall S, Strömberg B,
Holmström G, Thuresson AC, Annerén G and Bondeson ML: A severe form
of Noonan syndrome and autosomal dominant café-au-lait
spots-evidence for different genetic origins. Acta Paediatr.
98:693–698. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Spritz RA, Itin PH and Gutmann DH:
Piebaldism and neurofibromatosis type 1: Horses of very different
colors. J Invest Dermatol. 122:xxxiv–xxxv. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Duarte AF, Mota A, Baudrier T, Morais P,
Santos A, Cerqueira R, Tavares P and Azevedo F: Piebaldism and
neurofibromatosis type 1: Family report. Dermatol Online J.
16:112010.PubMed/NCBI
|
|
53
|
Chiu YE, Dugan S, Basel D and Siegel DH:
Association of Piebaldism, multiple café-au-lait macules, and
intertriginous freckling: Clinical evidence of a common pathway
between KIT and sprouty-related, ena/vasodilator-stimulated
phosphoprotein homology-1 domain containing protein 1 (SPRED1).
Pediatr Dermatol. 30:379–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Amyere M, Vogt T, Hoo J, Brandrup F, Bygum
A, Boon L and Vikkula M: KITLG mutations cause familial progressive
hyper- and hypopigmentation. J Invest Dermatol. 131:1234–1239.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Oiso N, Fukai K, Kawada A and Suzuki T:
Piebaldism. J Dermatol. 40:330–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Spritz RA: Molecular basis of human
piebaldism. J Invest Dermatol. 103:(Suppl 5). S137–S140. 1994.
View Article : Google Scholar
|
|
57
|
Angelo C, Cianchini G, Grosso MG, Zambruno
G, Cavalieri R and Paradisi M: Association of piebaldism and
neurofibromatosis type 1 in a girl. Pediatr Dermatol. 18:490–493.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Duarte A, Mota A, Baudrier T, Morais P,
Santos A, Cerqueira R, Tavares P and Azevedo F: Piebaldism and
neurofibromatosis: State of knowledge. Dermatol Online J.
19:172013.PubMed/NCBI
|
|
59
|
Wang ZQ, Si L, Tang Q, Lin D, Fu Z, Zhang
J, Cui B, Zhu Y, Kong X, Deng M, et al: Gain-of-function mutation
of KIT ligand on melanin synthesis causes familial progressive
hyperpigmentation. Am J Hum Genet. 84:672–677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Betz RC, Planko L, Eigelshoven S, Hanneken
S, Pasternack SM, Bussow H, Van Den Bogaert K, Wenzel J,
Braun-Falco M, Rutten A, et al: Loss-of-function mutations in the
keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet.
78:510–519. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pickup TL and Mutasim DF: Dowling-Degos
disease presenting as hypopigmented macules. J Am Acad Dermatol.
64:1224–1225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li M, Cheng R, Liang J, Yan H, Zhang H,
Yang L, Li C, Jiao Q, Lu Z, He J, et al: Mutations in POFUT1,
encoding protein O-fucosyltransferase 1, cause generalized
Dowling-Degos disease. Am J Hum Genet. 92:895–903. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Basmanav FB, Oprisoreanu AM, Pasternack
SM, Thiele H, Fritz G, Wenzel J, Größer L, Wehner M, Wolf S,
Fagerberg C, et al: Mutations in POGLUT1, encoding protein
O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos
disease. Am J Hum Genet. 94:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pérez B, Mechinaud F, Galambrun C, Ben
Romdhane N, Isidor B, Philip N, Derain-Court J, Cassinat B,
Lachenaud J, Kaltenbach S, et al: Germline mutations of the CBL
gene define a new genetic syndrome with predisposition to juvenile
myelomonocytic leukaemia. J Med Genet. 47:686–691. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Aoki Y, Niihori T, Banjo T, Okamoto N,
Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M, Ando T, et al:
Gain-of-function mutations in RIT1 cause Noonan syndrome, a
RAS/MAPK pathway syndrome. Am J Hum Genet. 93:173–180. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shah KN: The diagnostic and clinical
significance of café-au-lait macules. Pediatr Clin North Am.
57:1131–1153. 2010. View Article : Google Scholar : PubMed/NCBI
|