Introduction
Sulfur mustard (SM; 2,2′-dichlorodiethyl sulfide),
commonly known as mustard gas, is a potent bifunctional alkylating
agent (1), and was used as a
chemical weapon during World War I (1914–1918). Although more toxic
chemical warfare agents are currently available, mustard gas has
remained the chemical weapon of choice in modern warfare, as
evidenced by its use during the Iran-Iraq war between 1980 and 1988
(2). SM is a reactive chemical
causing skin, ocular and pulmonary damage (1). Among these, the primary target organ
of SM is the skin, which exhibits erythema, hyperpigmentation,
inflammation, blistering and severe necrosis (1). As no effective treatment for the
vesicant-inducing properties of SM has been established (3), the development of novel therapeutic
agents for SM-induced toxic symptoms is clinically urgent
considering the possibility of its use during future conflict.
Various molecules have been suggested to be involved
in the mechanism of SM-induced toxicity. Firstly, SM causes DNA
damage, which activates the poly (ADP-ribose) polymerase (PARP)
nuclear protein (1,4). The increased activation of PARP
depletes NAD+ and intracellular ATP levels, leading to
necrotic cell death. SM-induced DNA damage can also be caused by
its DNA adducts, which are formed predominantly at the N7-position
of guanine and the N1-/N3-position of adenine (1). These DNA adducts eventually cause
cell cycle arrest, leading to cell death. Secondly, SM induces
apoptotic cell death by increasing the expression levels of Fas
receptor, Fas ligands, tumor necrosis factor (TNF) receptor ligands
and TNF-α. SM also activates caspase-8 and its downstream caspases,
including caspase-3, −6 and −7 (1). Thirdly, endoplasmic reticulum stress
with changes in calcium homeostasis has also been reported to be
induced by SM through the modulation of calcium-calmodulin
signaling (1,5). SM can also alter signaling pathways,
including p38 mitogen-ativated protein (MAP) kinase and matrix
metalloproteases (MMPs). The SM-induced activation of p38 MAP
kinase is involved in cytokine release (6) and MMPs are important in SM-induced
skin blistering (1). Furthermore,
SM activates nuclear factor (NF)-κB signaling (7), and increases the generation of
reactive oxygen species (ROS) and nitric oxide, which depletes
glutathione (8). Therefore,
antioxidants have been used as therapeutic agents for SM-induced
toxicity (9–11). However, the therapeutic efficacy of
antioxidants is not clinically satisfactory and current treatments
are only supportive.
Historically, therapeutic agents were predominantly
obtained from various plants, and natural selection and competition
among species led to the synthesis of secondary metabolites with
marked biological activities (12). Caffeic acid (CA), an active
component of propolis, which has anti-inflammatory and
anticarcinogenic properties (13),
and inhibits 15-lipoxygenase (LOX) (14). Several other natural 15-LOX
inhibitors, including quercetin (15), a flavonoid found in a number of
vegetables and fruits, and morin hydrate, a yellow substance found
in oranges and guava (16), have
been reported previously.
LOX is an enzyme catalyzing the deoxygenation of
polyunsaturated fatty acids in lipids containing a cis,
cis-1,4-pentadiene structure, which generates the fatty acid,
hydroperoxide (17). LOX reactions
can modulate signaling pathways via the generation of leukotrienes
or 12-hydroxyeicosatetraenoic acid (12-HETE), but can also induce
structural or metabolic alterations inside the cell (17). In the present study, it was
demonstrated that CA, quercetin and morin hydrate not only reduced
SM-induced inflammatory cytokines via 15-LOX inhibition, but also
reduced SM-induced oxidative stress and the generation of
nitrate/nitrite.
Materials and methods
Materials
CA, tannic acid (TA), deferoxamine mesylate, trolox,
vitamin C, ellagic acid, N-acetyl-L-cysteine, quercetin, morin
hydrate, SB203580, PD146176 and 2′,7′-dichlorofluorescein diacetate
(DCF-DA) were purchased from Sigma-Aldrich; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Antibodies detecting
Thr183/Thr185-phosphorylated-c-jun N-terminal kinases (JNKs; cat.
no. 9255), JNKs (cat. no. 9252), Thr180/Tyr182-phosphorylated-p38
(cat. no. 4511), p38, Ser15-phosphorylated-p53 (cat. no. 9284), p53
(cat. no. 2524) and cyclooxygenase 2 (COX2; cat. no. 12282), all
purchased from Cell Signaling Biotechnology (Beverly, MA, USA), and
inducible nitric oxide synthase (iNOS; cat. no. ab3523; Abcam,
Cambridge, MA, USA). 12-LOX (cat. no. sc-32939) and 15-LOX (cat.
no. sc-67143) antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH; cat. no. MAB374)
antibody was purchased from EMD Millipore (Billerica, MA, USA).
Anti-mouse-horseradish peroxidase (HRP; cat. no. 115-036-003) and
anti-rabbit-HRP (cat. no. 111-035-003) antibodies were purchased
from Jackson Laboratory (Bar Harbor, ME, USA). The SM was provided
from the Agency for Defense Development (Daejeon, Korea).
Cell culture
Normal human epithelial keratinocytes (NHEKs) were
purchased from ZenBio, Inc. (Research Triangle Park, NC, USA). The
NHEKs were grown in KM-3 growth medium (ZenBio, Inc.) in 5%
CO2 at 37°C. All experiments were performed with NHEKs
at passages 3–5.
Measurements of TNF-α, interleukin-1β
(IL-1β) and 15-hydroxyeicosatetraenoic acid (15-HETE)
The NHEKs at 80% confluence were treated with 200 µM
of SM with or without antioxidants. After 18 h, the levels of
TNF-α, IL-1β and 15-HETE in the culture medium were measured using
TNF-α and IL-1β Human ELISA kits (AbFrontier, Seoul, Korea) and a
15-HETE ELISA kit (Abcam, Cambridge, MA, USA) according to the
manufacturer's protocols.
Western blot analysis
The NHEKs were lysed using RIPA buffer containing 50
mM Tris-Cl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium
deoxycholate, 0.1% SDS, protease inhibitors and phosphatase
inhibitors (Sigma-Aldrich; Thermo Fisher Scientific, Inc.) and
protein levels were examined using Protein Assay Dye reagent
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The proteins (50
µg) were separated by SDS-PAGE on 8–15% SDS polyacrylamide gels and
transferred onto PVDF membranes (Bio-Rad Laboratories, Inc.). The
membranes were blocked with the 5% skim milk for 1 h and incubated
with primary antibodies overnight at 4°C. The membranes were then
incubated with secondary antibodies for 1 h at room temperature.
The protein bands were detected on X-ray film using ECL Western
Blotting Detection reagents (GE Healthcare Life Sciences, Little
Chalfont, UK).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
cell viability assay
Cell viability was determined using an MTT assay.
Briefly, 5×104 NHEKs were grown overnight in 96-well plates, and
were treated with 0–800 µM SM for 24 h with or without antioxidants
(10 µM TA, 250 µM DFM, 250 µM Trolox, 100 mM vitamin C, 250 µM EA,
250 µM CA and 10 mM NAC). Following treatment, MTT solution was
added (0.5 mg/ml final concentration), and incubated for 4 h at
37°C with 5% CO2. The supernatant was discarded and 200
µl dimethyl sulfoxide was added. The production of solubilized
purple formazan crystals was quantified by exposure to a wavelength
of 540 nm.
Real-time cell electronic sensing
(RT-CES)
The NHEKs (5×104) were incubated in 16 plastic wells
with 16X microelectronic sensor devices. At 5 h post-cell
attachment, 200 µM SM with or without 250 µM CA, 100 µM quercetin
or 250 µM morin hydrate, was added. Data was collected every 3 min
using an RT-CES system (ACEA Biosciences, San Diego, CA, USA).
ROS measurement and nitrate/nitrite
assay
The NHEKs treated with 200 µM SM with or without
antioxidants (250 µM CA, 100 µM quercetin or 250 µM morin hydrate)
for 8 h, following which the NHEKs were washed with
phosphate-buffered saline (PBS) twice and incubated with 5 µM of
DCF-DA for 30 min at 37°C in the dark. Following incubation, the
NHEKs were washed with PBS twice and fluorescence intensity was
detected with an excitation wavelength of 492 nm and emission of
530 nm. Total nitrate/nitrite levels in the cell culture medium
were quantified using a Nitrate/Nitrite Colorimetric Assay kit
(Cayman Chemical Co., Ann Arbor, MI, USA) according to the
manufacturer's protocol.
Statistical analysis
All experiments were repeated at least three times
independently, and values are presented as the mean ± standard
error of the mean. Statistical significance was calculated using
Student's t-test using Prism 6.0 software (GraphPad, San Diego, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
CA alleviates SM-induced
cytotoxicity
To confirm the toxicity of SM in NHEK cells, various
SM concentrations were used to treat NHEKs for 24 h. As expected,
SM treatment reduced cell viability in a dose-dependent manner, and
cell viability was ~50% at the 200 µM concentration of SM (Fig. 1A). To identify potential
therapeutic candidates for SM-induced cytotoxicity, several
antioxidants were incubated with SM. Among these, CA (250 µM)
showed a protective effect against SM-induced cytotoxicity
(Fig. 1B). In addition, CA
treatment at concentrations >250 µM recovered cell viability in
a dose-dependent manner (Fig. 1C).
As SM has previously been reported to increase the phosphorylation
of p38 MAP kinase and p53 (1,18),
the present study examined the MAP kinase and p53 pathways
following treatment with SM and CA. SM treatment increased the
phosphorylation of JNK1/2, p38 and p53, as reported previously
(1,18). CA treatment decreased the
SM-induced phosphorylation of p38 and p53, and increased the
phosphorylation of JNK 1/2 (Fig.
1D).
 | Figure 1.CA treatment reduces SM-induced cell
death. NHEKs were treated with SM for 24 h. Cell viability assays
were performed with (A) various SM concentrations (n=4) and with
(B) 200 µM SM and the respective antioxidants (n=4). (C) Various
concentrations of CA were co-incubated with SM, and a cell
viability assay was performed (n=4). (D) Western blots of NHEK
lysates following 8 h treatment with SM with or without CA. The
values are expressed as the mean ± standard error of the mean.
**P<0.01; ***P<0.001. Four independent experiments were
performed. NHEKs, normal human epidermal keratinocytes; SM, sulfur
mustard; TA, tannic acid; DFM, deferoxamine methylate; EA, elagic
acid; CA, caffeic acid; NAC, N-acetyl cysteine; JNK, c-Jun
N-terminal kinase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase;
p-, phosphorylated; O.D., optical density. |
CA reduces SM-induced cytotoxicity via
the inhibition of LOX
To determine whether the p38 pathway is involved in
the protective effect of CA on SM-induced cytotoxicity, a p38
inhibitor (SM203580) was co-incubated with SM and CA. p38
inhibition by SM203580 did not affect either SM-induced
cytotoxicity or CA-induced cell recovery (Fig. 2A). As CA is also known as a LOX
inhibitor (14), the present study
examined whether the expression levels of LOX are altered upon SM
treatment. Treatment with SM elevated the protein expression levels
of 12- and 15-LOX, which were abrogated by CA treatment (Fig. 2B). In addition, treatment with the
specific 15-LOX inhibitor, PD146176, significantly increased
SM-treated cell viability, similar to the effects of CA (Fig. 2C), which confirmed the role of
15-LOX in SM-induced cytotoxicity. The RT-CES technique was used to
confirm the improved cell viability by PD146176, the results of
which also showed the protective effect of the 15-LOX inhibitor
(PD146176) against SM (Fig.
2D).
Other plant-derived LOX inhibitors,
morin hydrate and quercetin also reduce SM-induced
cytotoxicity
To confirm whether the LOX pathway is involved in
SM-induced cytotoxicity, other naturally obtained 15-LOX
inhibitors, including morin hydrate and quercetin (19), were co-incubated with SM. Treatment
of cells with morin hydrate and quercetin partially recovered
SM-induced cell death (Fig. 3A).
Similarly, the RT-CES data showed delayed cell death upon morin
hydrate and quercetin treatment (Fig.
3B). The reductions in the protein levels of 12- and 15-LOX by
incubation with morin hydrate and quercetin were confirmed using
western blot analysis (Fig. 3C).
Furthermore, morin hydrate and quercetin reduced the
phosphorylation of p38 MAP kinase (Fig. 3C). However, PD146176 and SB203580
did not affect the expression levels of 12-LOX or 15-LOX, and only
SB203580 reduced SM-induced p38 phosphorylation (Fig. 3D).
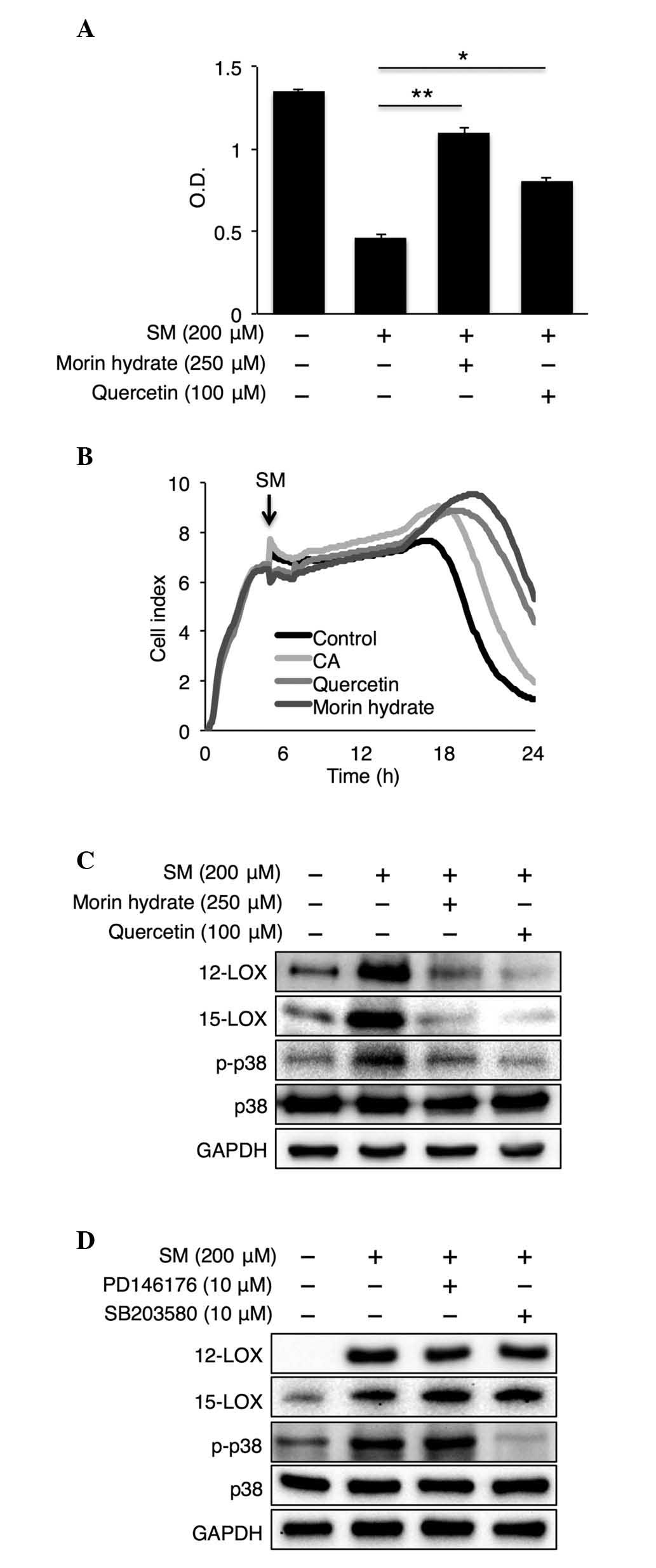 | Figure 3.Natural 15-LOX inhibitors, quercetin
and morin hydrate, have protective effects against SM-induced
toxicity. (A) Cell viability assay following co-reatment with SM
(200 µM) and quercetin or morin hydrate (n=4). (B) Real-time cell
electronic sensing data of co-treatment with SM (200 µM) and CA,
quercetin or morin hydrate. Experiments were repeated at least four
times with similar results. (C) Representative western blots of
NHEK lysates following co-treatment with SM (200 µM) and quercetin
or morin hydrate, and (D) following co-treatment with SM (200 µM)
with PD146176 or SB203580. Data are expressed as the mean ±
standard error of the mean. *P<0.05 and **P<0.01. Four
independent experiments are shown. NHEKs, normal human epidermal
keratinocytes; CA, caffeic acid; SM, sulfur mustard; LOX,
lipoxygenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Con
control; O.D., optical density; p-, phosphorylated. |
LOX inhibition reduces SM-induced
levels of TNF-α, but not IL-1β
SM is also reported to elevate inflammatory
cytokines, including TNF-α and IL-1 β (6), thus, the present study measured the
levels of TNF-α and IL-1β upon SM treatment with LOX inhibitors
(CA, quercetin, morin hydrate and PD146176) or the p38 inhibitor
(SB203580). p38 inhibition by SB203580 reduced the levels of TNF-α
and IL-1β produced by SM, whereas the LOX inhibitors exerted
different effects on cytokine production depending on their
properties on the p38 pathway (Fig. 4A
and B). Although all four LOX inhibitors reduced TNF-α levels,
only CA, quercetin and morin hydrate, which inhibited p38 MAP
kinase (Fig. 3C), reduced the
levels of IL-1β produced by SM. PD146176, which did not reduce
SM-induced p38 phosphorylation (Fig.
3D), did not reduce the levels of SM-induced IL-1β. All four
LOX inhibitors decreased the SM-induced production of 15-HETE, as
expected, which indicated effective LOX inhibition (Fig. 4C).
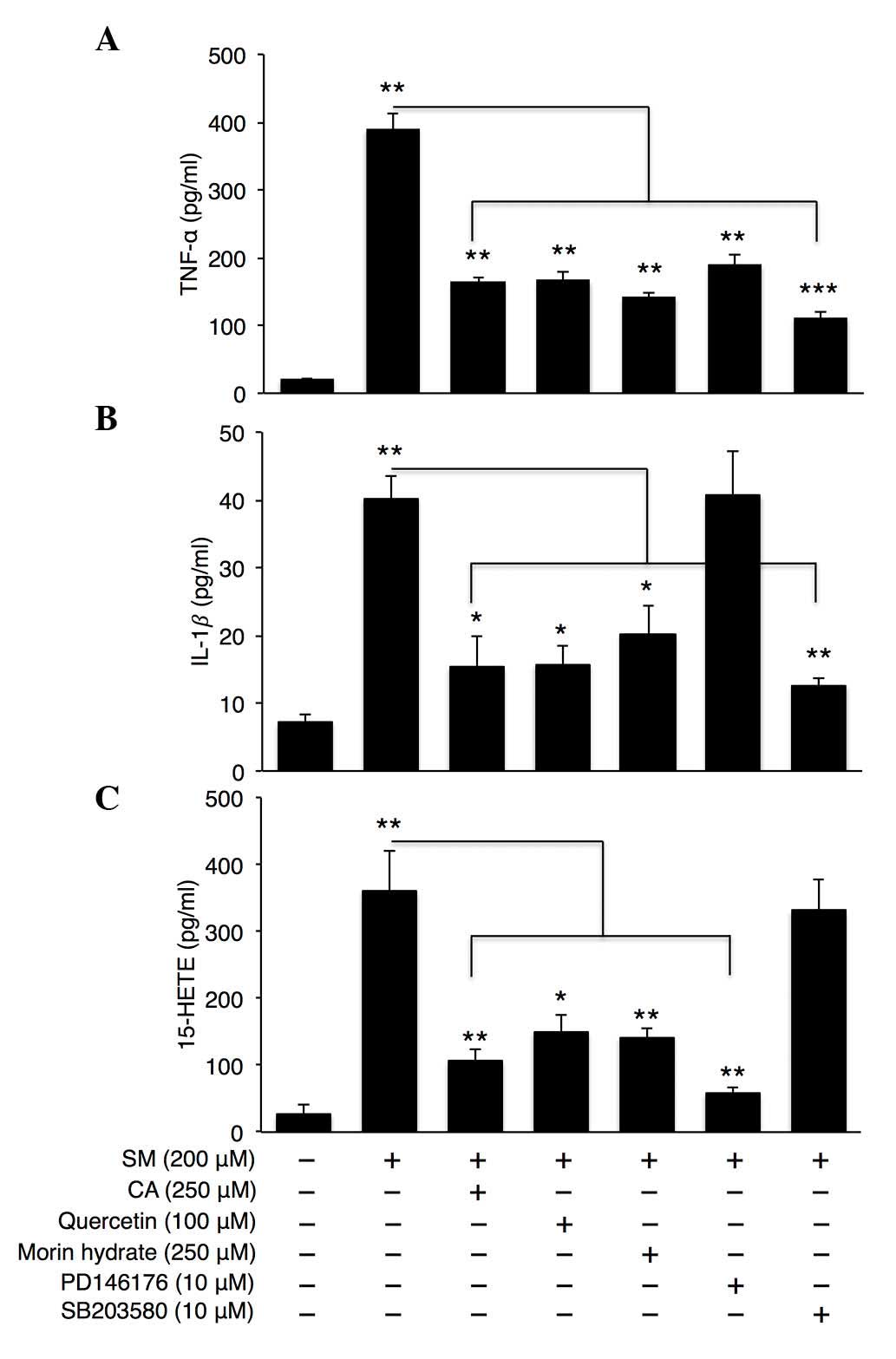 | Figure 4.15-LOX inhibition reduces levels of
TNF-α and 15-HETE inflammatory cytokines. Normal human epidermal
keratinocytes were treated with SM (200 µM), antioxidants (CA,
quercetin or morin hydrate) and specific inhibitors of p38
(SB203580) and 15-LOX pathways (PD146176) for 24 h. Expression
levels of (A) TNF-α, (B) IL-1β and (C) 15-HETE were measured (n=4).
Data are expressed as the mean ± standard error of the mean
*P<0.05, **P<0.01 and ***P<0.001. Four independent
experiments are shown. LOX, lipoxygenase; SM, sulfur mustard; CA,
caffeic acid; TNF-α, tumor necrosis factor-α; IL-1β, interleukin
−1β, 15-HETE, hydroxyeicosatetraenoic acid. |
CA, quercetin and morin hydrate reduce
the expression levels of COX2 and iNOS
Finally, the present study examined COX2 and iNOS
signaling in SM-treated NHEKs. CA, quercetin and morin hydrate
reduced the expression levels of COX2 and iNOS (Fig. 5A), whereas SB203580 and PD146176
did not affect the levels of COX2 or iNOS (Fig. 5B). In addition, CA, quercetin and
morin hydrate attenuated nitrate/nitrite and ROS generation
(Fig. 5C and D). From these
results, it was concluded that CA, quercetin and morin hydrate
reduced COX2, iNOS and the generation of oxidative stress
irrespective of the p38 and LOX pathway.
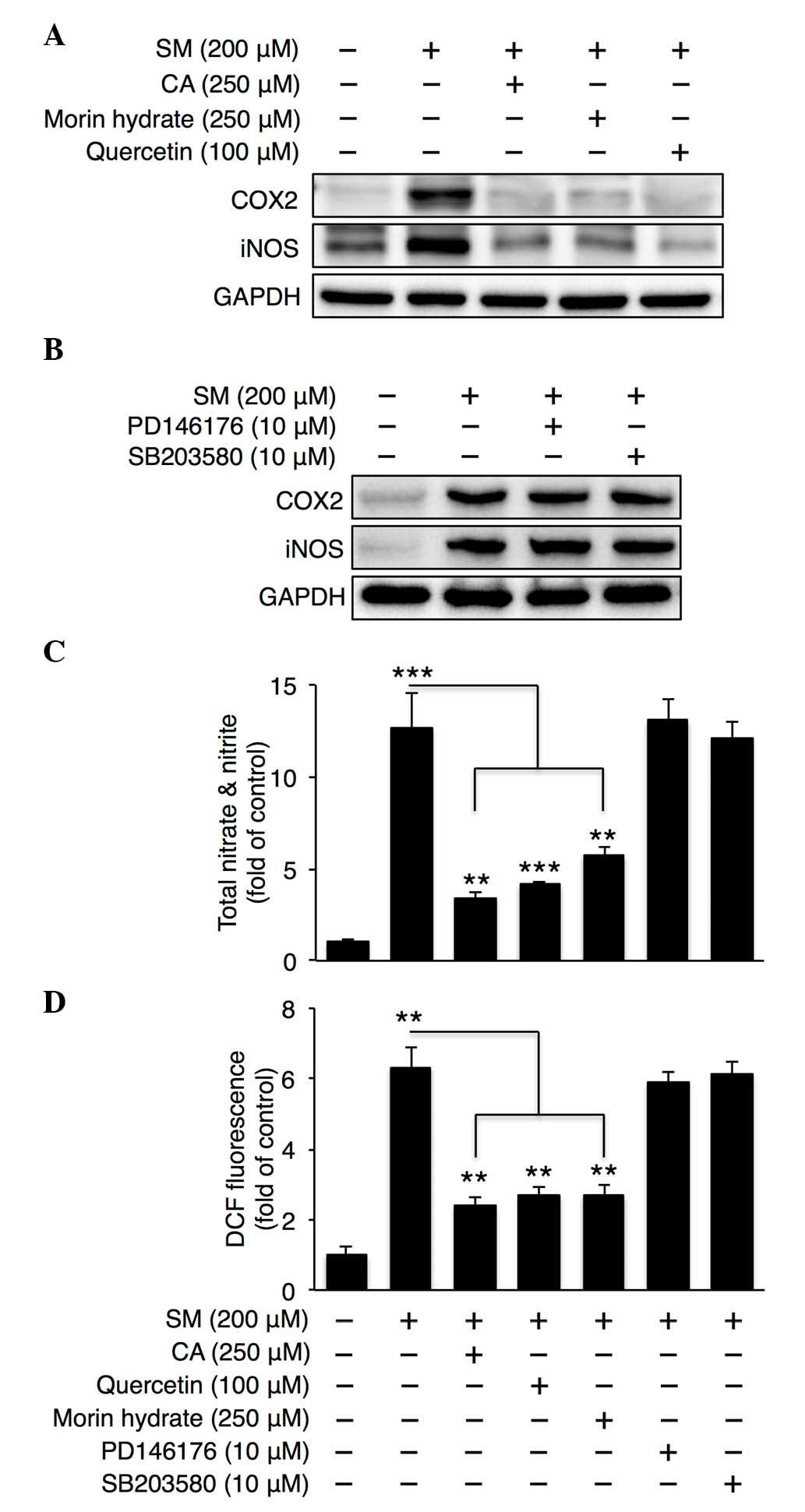 | Figure 5.CA, quercetin and morin hydrate
reduce ROS and nitrate/nitrite generation by SM. Representative
western blots of normal human epidermal keratinocyte lysates
following treatment with SM (200 µM) and (A) CA, quercetin or morin
hydrate, or (B) SB203580 or PD146176. (C) Total nitrate/nitrite and
(D) ROS generation were measured following co-treatment with SM
(200 µM) and CA or quercetin or morin hydrate antioxidants (n=4).
Data are expressed as the mean ± standard error of the mean.
**P<0.01 and ***P<0.001. Four independent experiments are
shown. CA, caffeic acid; SM, sulfur mustard; LOX, lipoxygenase;
ROS, reactive oxygen species; COX2, cyclooxygenase 2; GAPDH,
glyceraldehyde-3-phosphate dehydrogenase; iNOS, inducible nitric
oxide synthase; DCF, 2′,7′-dichlorofluorescein. |
Discussion
Several mechanisms, including inflammation and
oxidative stress, have been reported to be involved in SM-induced
cytotoxicity (6,8,20–22).
For example, SM induces the production of inflammatory cytokines
via p38 MAP kinase (6). SM also
accelerates the release of arachidonic acids (21), which can be converted to
prostaglandins and leukotrienes (23). SM exposure can also activate
several signaling pathways, including the NF-κB and p53 pathway
(1,22). In the present study, it was found
that the administration of CA, which has a LOX inhibitory effect,
reduced the inflammatory response and cytotoxicity induced by SM.
CA treatment decreased the phosphorylation of p38 MAP kinase and
the elevation of LOX, resulting in decreased production of
inflammatory cytokines and cytotoxicity. Furthermore, all LOX
inhibitors, including quercetin, morin hydrate and PD146176,
partially protected the NHEKs from SM-induced cell death. Several
previous studies have reported the involvement of LOX in cell death
(24–27). In fibroblasts, 12-LOX activation
leads to apoptosis, and the 12-LOX product,
12-hydroperoxyeicosatetraenoic acid, directly induces apoptosis in
CHO-AT1a cells (25). In addition,
12-LOX activation is important in oxidative stress-induced
oligodendrocyte death (26), and
the activation of neuronal 12-LOX leads to the production of
peroxides and influx of Ca2+, ultimately leading to cell
death (27). Additionally,
12/15-LOX-derived lipid peroxidation triggers apoptosis-inducing
factor-mediated cell death (24).
The results of the present study suggested that the LOX pathway was
also involved in SM-induced cell death, and that inhibiting the LOX
pathway offers a potential therapeutic target for SM-induced
toxicity.
A crucial role of the 5-LOX/12-LOX/15-LOX pathway in
regulating the production of TNF-α and TNF-α-mediated inflammation
has been previously reported (28–30).
In accordance with these previous studies, the present study showed
that CA, quercetin and morin hydrate decreased the production of
TNF-α, not only by the p38 pathway, but also through the inhibition
of 15-LOX. However, unlike the production of TNF-α, inhibition of
the p38 pathway, but not of 15-LOX, reduced the production of
IL-1β. Furthermore, CA, quercetin and morin hydrate reduced the
generation of ROS and nitrate/nitrite. As neither p38 inhibition
nor 15-LOX inhibition affected the generation of ROS or
nitrate/nitrite, these effects may be derived from their scavenging
effects of free radicals (31–33).
Although the activation of MAP kinases, including
p38 and JNK-1/-2, was induced by SM, CA treatment reduced the
phosphorylation of p38 MAP kinase, but not JNK-1/-2. In a previous
study, CA was reported to paradoxically increase the
phosphorylation of JNK (34). As
CA treatment did not reduce SM-induced JNK phosphorylation, the
precise role of JNK activation in SM-induced cytotoxicity remains
to be elucidated.
Various mechanisms involved in SM-induced toxicity
have been reported (1) and current
knowledge of the mechanism underlying SM-induced toxicity is
increasing. Although the development of therapeutic agents against
SM is complex due to the complexity of the mechanisms involved, a
combinational approach may lead to an effective treatment strategy
for SM-induced toxicity. The present study suggested that natural
LOX inhibitors, including CA, quercetin and morin hydrate, can be
used as early therapeutic agents for SM-induced toxicity by
reducing the early inflammatory responses, oxidative stress and
expression of COX2/iNOS induced by SM (Fig. 6).
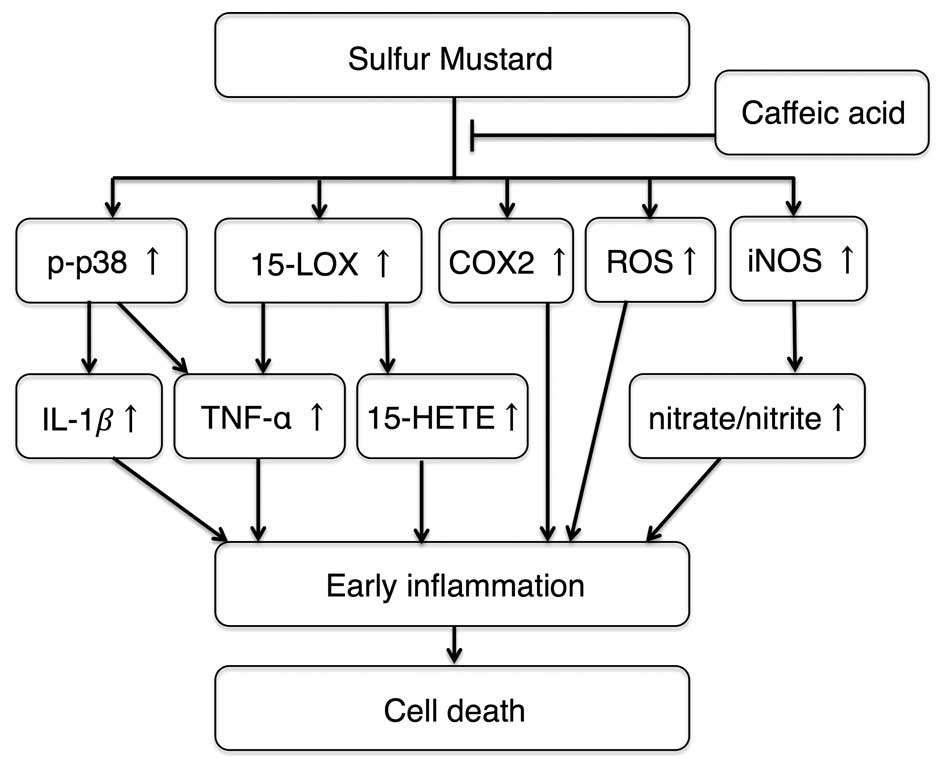 | Figure 6.Schematic showing the effects of CA
in sulfur mustard-induced toxicity. CA treatment reduces p38
phosphorylation, which reduces the levels of TNF-α and IL-1β.
15-LOX inhibition by CA attenuates the production of TNF-α and
15-HETE. CA also reduces the levels of COX2 and iNOS, and decreases
ROS and nitrate/nitrite generation. These effects decrease
SM-induced inflammation and cell death. CA, caffeic acid; LOX,
lipoxygenase; COX2, cyclooxygenase 2; ROS, reactive oxygen species;
iNOS, inducible nitric oxide synthase; IL-1β, interleukin −1β,
TNF-α, tumor necrosis factor-α; 15-HETE, hydroxyeicosatetraenoic
acid. |
Acknowledgements
All experiments were performed between 2009 and
2011. Joo-Won Park was supported by the Health Technology R&D
project (grant no. HI14C2445) of the Ministry of Health and
Welfare, Republic of Korea. Woo-Jae Park was supported by a Gachon
University Research Grant of 2015 (grant no. GCU-2015-5112). Shin
Kim was supported by the National Research Foundation of Korea
Grant funded by the Korean Government (grant. no.
2014R1A5A2010008)
Glossary
Abbreviations
Abbreviations:
|
SM
|
sulfur mustard
|
|
CA
|
caffeic acid
|
|
COX2
|
cyclooxygenase 2
|
|
HETE
|
hydroxyeicosatetraenoic acid
|
|
IL-1β
|
interleukin-1β
|
|
iNOS
|
inducible nitric oxide synthase
|
|
LOX
|
lipoxygenase
|
|
ROS
|
reactive oxygen species
|
|
TNF-α
|
tumor necrosis factor-α
|
|
RT-CES
|
real-time cell electronic sensing
|
References
|
1
|
Kehe K, Balszuweit F, Steinritz D and
Thiermann H: Molecular toxicology of sulfur mustard-induced
cutaneous inflammation and blistering. Toxicology. 263:12–19. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ries C, Popp T, Egea V, Kehe K and Jochum
M: Matrix metalloproteinase-9 expression and release from skin
fibroblasts interacting with keratinocytes: Upregulation in
response to sulphur mustard. Toxicology. 263:26–31. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McClintock SD, Hoesel LM, Das SK, Till GO,
Neff T, Kunkel RG, Smith MG and Ward PA: Attenuation of half sulfur
mustard gas-induced acute lung injury in rats. J Appl Toxicol.
26:126–131. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kehe K, Raithel K, Kreppel H, Jochum M,
Worek F and Thiermann H: Inhibition of poly (ADP-ribose) polymerase
(PARP) influences the mode of sulfur mustard (SM)-induced cell
death in HaCaT cells. Arch Toxicol. 82:461–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Simbulan-Rosenthal CM, Ray R, Benton B,
Soeda E, Daher A, Anderson D, Smith WJ and Rosenthal DS: Calmodulin
mediates sulfur mustard toxicity in human keratinocytes.
Toxicology. 227:21–35. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dillman JF III, McGary KL and Schlager JJ:
An inhibitor of p38 MAP kinase downregulates cytokine release
induced by sulfur mustard exposure in human epidermal
keratinocytes. Toxicol In Vitro. 18:593–599. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rebholz B, Kehe K, Ruzicka T and Rupec RA:
Role of NF-kappaB/RelA and MAPK pathways in keratinocytes in
response to sulfur mustard. J Invest Dermatol. 128:1626–1632. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pal A, Tewari-Singh N, Gu M, Agarwal C,
Huang J, Day BJ, White CW and Agarwal R: Sulfur mustard analog
induces oxidative stress and activates signaling cascades in the
skin of SKH-1 hairless mice. Free Radic Biol Med. 47:1640–1651.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Paromov V, Suntres Z, Smith M and Stone
WL: Sulfur mustard toxicity following dermal exposure: Role of
oxidative stress and antioxidant therapy. J Burns Wounds.
7:e72007.PubMed/NCBI
|
|
10
|
Kumar O, Sugendran K and Vijayaraghavan R:
Protective effect of various antioxidants on the toxicity of
sulphur mustard administered to mice by inhalation or percutaneous
routes. Chem Biol Interact. 134:1–12. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Naghii MR: Sulfur mustard intoxication,
oxidative stress, and antioxidants. Mil Med. 167:573–575.
2002.PubMed/NCBI
|
|
12
|
Gamaro GD, Suyenaga E, Borsoi M, Lermen J,
Pereira P and Ardenghi P: Effect of rosmarinic and caffeic acids on
inflammatory and nociception process in rats. ISRN Pharmacol.
2011:4516822011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Natarajan K, Singh S, Burke TR Jr,
Grunberger D and Aggarwal BB: Caffeic acid phenethyl ester is a
potent and specific inhibitor of activation of nuclear
transcription factor NF-kappa B. Proc Natl Acad Sci USA.
93:9090–9095. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sud'ina GF, Mirzoeva OK, Pushkareva MA,
Korshunova GA, Sumbatyan NV and Varfolomeev SD: Caffeic acid
phenethyl ester as a lipoxygenase inhibitor with antioxidant
properties. FEBS Lett. 329:21–24. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sultana B and Anwar F: Flavonols
(kaempeferol, quercetin, myricetin) contents of selected fruits,
vegetables and medicinal plants. Food Chem. 108:879–884. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rattanachaikunsopon P and Phumkhachorn P:
Bacteriostatic effect of flavonoids isolated from leaves of Psidium
guajava on fish pathogens. Fitoterapia. 78:434–436. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brash AR: Lipoxygenases: Occurrence,
functions, catalysis, and acquisition of substrate. J Biol Chem.
274:23679–23682. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Minsavage GD and Dillman JF III:
Bifunctional alkylating agent-induced p53 and nonclassical nuclear
factor kappaB responses and cell death are altered by caffeic acid
phenethyl ester: A potential role for antioxidant/electrophilic
response-element signaling. J Pharmacol Exp Ther. 321:202–212.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sadik CD, Sies H and Schewe T: Inhibition
of 15-lipoxygenases by flavonoids: Structure-activity relations and
mode of action. Biochem Pharmacol. 65:773–781. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Black AT, Joseph LB, Casillas RP, Heck DE,
Gerecke DR, Sinko PJ, Laskin DL and Laskin JD: Role of MAP kinases
in regulating expression of antioxidants and inflammatory mediators
in mouse keratinocytes following exposure to the half mustard,
2-chloroethyl ethyl sulfide. Toxicol Appl Pharmacol. 245:352–360.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lefkowitz LJ and Smith WJ: Sulfur
mustard-induced arachidonic acid release is mediated by
phospholipase D in human keratinocytes. Biochem Biophys Res Commun.
295:1062–1067. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gerecke DR, Chen M, Isukapalli SS, Gordon
MK, Chang YC, Tong W, Androulakis IP and Georgopoulos PG:
Differential gene expression profiling of mouse skin after sulfur
mustard exposure: Extended time response and inhibitor effect.
Toxicol Appl Pharmacol. 234:156–165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Folco G and Murphy RC: Eicosanoid
transcellular biosynthesis: From cell-cell interactions to in vivo
tissue responses. Pharmacol Rev. 58:375–388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Seiler A, Schneider M, Förster H, Roth S,
Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et
al: Glutathione peroxidase 4 senses and translates oxidative stress
into 12/15-lipoxygenase dependent- and AIF-mediated cell death.
Cell Metab. 8:237–248. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gu J, Liu Y, Wen Y, Natarajan R, Lanting L
and Nadler JL: Evidence that increased 12-lipoxygenase activity
induces apoptosis in fibroblasts. J Cell Physiol. 186:357–365.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Li J, Follett PL, Zhang Y,
Cotanche DA, Jensen FE, Volpe JJ and Rosenberg PA: 12-Lipoxygenase
plays a key role in cell death caused by glutathione depletion and
arachidonic acid in rat oligodendrocytes. Eur J Neurosci.
20:2049–2058. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li Y, Maher P and Schubert D: A role for
12-lipoxygenase in nerve cell death caused by glutathione
depletion. Neuron. 19:453–463. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martínez-Clemente M, Ferré N,
González-Périz A, López-Parra M, Horrillo R, Titos E,
Morán-Salvador E, Miquel R, Arroyo V, Funk CD and Clària J:
5-lipoxygenase deficiency reduces hepatic inflammation and tumor
necrosis factor alpha-induced hepatocyte damage in
hyperlipidemia-prone ApoE-null mice. Hepatology. 51:817–827. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wen Y, Gu J, Chakrabarti SK, Aylor K,
Marshall J, Takahashi Y, Yoshimoto T and Nadler JL: The role of
12/15-lipoxygenase in the expression of interleukin-6 and tumor
necrosis factor-alpha in macrophages. Endocrinology. 148:1313–1322.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schade UF, Ernst M, Reinke M and Wolter
DT: Lipoxygenase inhibitors suppress formation of tumor necrosis
factor in vitro and in vivo. Biochem Biophys Res Commun.
159:748–754. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang R, Kang KA, Piao MJ, Maeng YH, Lee
KH, Chang WY, You HJ, Kim JS, Kang SS and Hyun JW: Cellular
protection of morin against the oxidative stress induced by
hydrogen peroxide. Chem Biol Interact. 177:21–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dugas AJ, Castañeda-Acosta J, Bonin GC,
Price KL, Fischer NH and Winston GW: Evaluation of the total
peroxyl radical-scavenging capacity of flavonoids:
Structure-activity relationships. J Nat Prod. 63:327–331. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen YJ, Shiao MS and Wang SY: The
antioxidant caffeic acid phenethyl ester induces apoptosis
associated with selective scavenging of hydrogen peroxide in human
leukemic HL-60 cells. Anticancer Drugs. 12:143–149. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi K and Choi C: Differential regulation
of c-Jun N-terminal kinase and NF-kappaB pathway by caffeic acid
phenethyl ester in astroglial and monocytic cells. J Neurochem.
105:557–564. 2008. View Article : Google Scholar : PubMed/NCBI
|















