Introduction
Down syndrome (DS) or Down's syndrome, also known as
trisomy 21, is an autosomal abnormality induced by an extra copy of
chromosome 21 and is the most common birth defect among children
worldwide (1,2). Children with DS usually have severe
mental retardation (3) and delayed
development (4), and are prone to
gastrointestinal malformations (5). In children, almost 50% of DS cases
are accompanied with congenital heart disease (6), and the risk of developing acute
leukemia is 20 times higher than that of the normal population
globally (7). In addition,
patients with DS generally have shorter life expectancy (8,9).
DS is the most common cause of mental retardation
and malformation in newborns. During meiosis, chromosome 21 in the
egg does not separate, therefore, an extra copy of chromosome 21 is
produced (10). When the sperm and
the egg fuse, the embryo has 47 chromosomes, with three copies of
chromosome 21 (11). An extra
chromosome 21 leads to the overexpression of its genes, causing
nerve dysfunction in vivo, and affecting the normal growth
and development of children (12).
At present, prenatal diagnosis is the optimal approach in
preventing DS, however, there are no effective drugs for treatment
of the disease. Thus, it is important to investigate the molecular
mechanisms of DS.
Previous studies have suggested that the elevated
gene expression of human chromosome 21 (HSA21) is
responsible for specific aspects of the DS phenotype. Arron et
al (13) showed that certain
characteristics of the DS phenotype can be associated with the
increased expressions of two HSA21 genes, namely those
encoding the transcriptional activator, regulator of calcineurin 1
(DSCR1-RCAN1), and the protein kinase, dual-specificity
tyrosine phosphorylation-regulated kinase (DYRK)1A. The
overexpression of a number of HSA21 genes, including
DYRK1a, synaptogenin 1 and single-minded homolog 2, results
in learning and memory defects in mouse models, suggesting that
trisomy of these genes may contribute to learning disability in
patients with DS (14,15).
The abnormal copy number of chromosome 21 is the
primary genetic characteristics of DS. Therefore, the present study
applied a variety of bioinformatics tools to determine the genetic
fragments in chromosome 21. The methylated sites in
bisulfite-sequencing (seq) data were detected, differentially
methylated regions between DS and control samples were determined,
and the adjacent genes of differential DNA methylation regions were
identified. Subsequently, the functions of the abnormal
demethylated genes were predicted using Gene Ontology (GO)
enrichment analyses. The differentially expressed genes (DEGs)
between the DS and control samples were screened. Furthermore, the
interactions/associations between the proteins encoded by selected
genes were determined, and a protein-protein interaction (PPI)
network was constructed. The present study aimed to identify the
key genes involved in DS, and may be able to establish the
theoretical foundation for the targeted therapy of DS.
Materials and methods
Data sources
The bisulfite-seq data GSE42144 deposited by Jin
et al (16) was downloaded
from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) of the National
Center for Biotechnology Information, which was based on the
platform of the Illumina Genome Analyzer IIx (Illumina, Inc., San
Diego, CA, USA). GSE42144 included three placental samples
(GSM1032059, GSM1032060 and GSM1032061) from patients with DS and
three normal control samples (GSM1032070, GSM1032071 and
GSM1032072). According to the quality control results of RNA-seq
data, two RNA-seq data from patients with DS (GSM1033476 and
GSM1033478) and five RNA-seq data from normal control samples
(GSM1033470, GSM1033471, GSM1033472, GSM1033473 and GSM1033474)
were also used in the present study.
Alignment of bisulfite-seq data and
detection of DNA methylation
For all bisulfite-seq data, Bismark (www.bioinformatics.bbsrc.ac.uk/projects/bismark/)
(17) and Bowtie 2 (bowtie-bio.sourceforge.net/bowtie2/index.shtml)
(18) software were applied to
perform read alignment, analyze methylated DNA signaling and output
cytosine methylation sites in the genome. All parameters were set
at default values.
Differential DNA methylation and
corresponding adjacent gene analysis
The BiSeq tool (19) was used to determine differentially
methylated regions between the placenta samples of patients with DS
and normal control samples. The false discovery rate of each
significant differentially methylated CpG cluster was ≤0.1. The
methylated CpG clusters with a length of <100 bp were merged and
defined as a differentially methylated DNA region. In addition, the
length of each differentially methylated DNA region was required to
be ≥50 bp. If the distance between the center of the differentially
methylated DNA region and the transcription start site of a
specific gene ranged between 3,000 and 500 bp, the differentially
methylated DNA region was considered to have the potential to
affect the gene, and this gene was defined as an adjacent gene of
the differentially methylated DNA region.
Alignment of RNA-seq data and
calculation of gene expression
Tophat (4) software
was used to perform read alignment, with the University of
California Santa Cruz (genome.ucsc.edu) hg19 genome sequences as a reference.
For read alignment, up to two base mismatches were permitted in one
read. Only the reads which mapped to specific genome locations were
retained for further analysis. The other parameters were set to the
defaults. On combining with the Refseq gene annotations, the
transcripts were assembled and gene expression values were
calculated using Cufflinks and Cuffdiff tools (5). The calculated gene expression values
were based on the fragments per kilobase of transcript per million
fragments mapped method (20).
Analysis of DEGs
The paired t-test (21) was used to identify DEGs between the
DS and control samples. P<0.01 and |log2fold-change|≥2 were used
as the cut-off criteria.
Function annotation of the adjacent
genes
The Database for Annotation, Visualization, and
Integrated Discovery (22) was
used to perform GO enrichment analysis for the adjacent genes of
the differentially methylated DNA regions. The GO terms were
classified into biological process, molecular function and cellular
component categories. P<0.05 was used as the cut-off
criterion.
PPI network construction
The interaction associations of the proteins encoded
by selected genes were determined using the Search Tool for the
Retrieval of Interacting Genes (STRING) database (23). All parameters were set to defaults.
A PPI network was then constructed using Cytoscape (24).
Results
Identification of differential DNA
methylation regions in DS
Based on the Bisulfite-seq data, a total of 74 CpG
regions had significant differential DNA methylation between the DS
and normal samples, including 68 demethylated regions, accounting
for 92%, and six regions with higher levels of methylation,
compared with those of the normal samples.
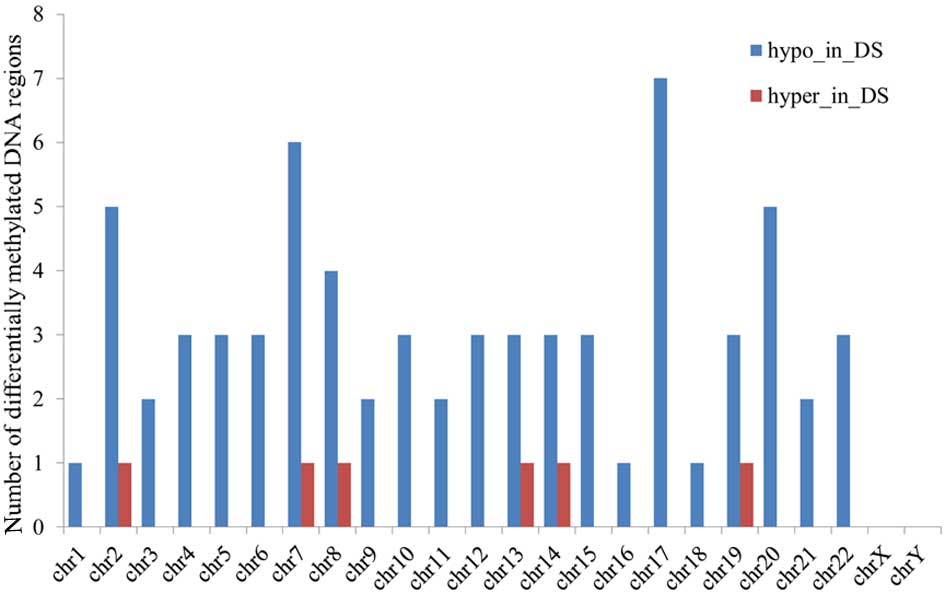
For a single chromosome, the majority of the
abnormal DNA methylation regions were detected in chromosomes 7 and
17, showing a total of seven aberrantly methylated DNA regions. In
chromosome 21, five abnormal demethylated DNA regions were found
(Fig. 1).
Identification of adjacent genes of
the differentially methylated DNA
Compared with the control samples, a total of 43
adjacent (protein-coding) genes were identified in the DS samples
with demethylated promoter regions and one adjacent gene,
chromosome 19 open reading frame 80, which is known to be located
in chromosome 19, was identified with upregulated methylation in
its promoter region (Table I).
 | Table I.Number of adjacent genes with
differentially methylated DNA regions in Down syndrome samples. |
Table I.
Number of adjacent genes with
differentially methylated DNA regions in Down syndrome samples.
| Methylation | Protein-coding
genes (n) | Noncoding genes
(n) |
|---|
| Hypomethylated | 43 | 0 |
|
Hypermethylated | 1 | 0 |
In the autosomal chromosomes, there were six
DS-associated genes with demethylated promoter regions in
chromosome 17. The number of abnormal genes in other chromosomes
ranged between one and three. No genes were found to be affected by
abnormal DNA demethylation in the sex chromosomes.
The distributions of the genes on chromosomes are
listed in Table II. Among these,
only Runt-related transcription factor 1 (RUNX1) was found
to be located on chromosome 21, and the demethylation of the
promoter region of this gene was significant (Table II).
| Table II.Distribution of adjacent genes with
demethylated regions in DS sample chromosomes. |
Table II.
Distribution of adjacent genes with
demethylated regions in DS sample chromosomes.
| Chromosome | n | Gene |
|---|
| 1 | 2 | LOC730144,
EIF1 |
| 2 | 3 | ATG4B, LRRFIP1,
STRADB |
| 3 | 1 | EXOG |
| 4 | 1 | FRYL |
| 5 | 3 | BOD1, B4GALT7,
BRD9 |
| 6 | 2 | MAP3K4, ID4 |
| 7 | 4 | PAXIP1, POLM,
FAM131B, AGAP3 |
| 8 | 2 | ATAD2, REEP4 |
| 9 | 2 | BARX1,
KIAA1539 |
| 10 | 2 | OTUD1, GPRIN2 |
| 11 | 1 | SLC1A2 |
| 12 | 1 | ALDH2 |
| 13 | 3 | HMGB1, HMGB1L10,
FARP1 |
| 14 | 1 | RTN1 |
| 15 | 2 | TYRO3, PCSK6 |
| 16 | 1 | FA2H |
| 17 | 6 | UTP18, LOC730144,
C17ORF56, EIF1, TIMP2, MRM1 |
| 18 | 1 | ABHD3 |
| 20 | 2 | CBFA2T2,
C20ORF20 |
| 21 | 1 | RUNX1 |
| 22 | 3 | HMGB1, OSBP2,
HMGB1L10 |
Functional enrichment of abnormal
demethylated genes
The present study subsequently analyzed the
functions of the 43 abnormally demethylated genes. Combined with GO
functional annotation, the five genes, high mobility group box
(HMGB)1, HMGB1L10, inhibitor of DNA binding 4
(ID4), leucine-rich repeat flightless-interacting protein 1
and core-binding factor, Runt domain, α subunit 2; translocated to,
2 (CBFA2T2) were found to be involved in the biological
process of negative regulation of transcription, whereas the three
genes, BARX homeobox 1, DNA polymerase mu and RUNX1, were
associated with immune system development. In addition, the present
study found that solute carrier family 1 member 2, ID4 and
tissue inhibitor of matrix metalloproteinase 2 were predominantly
involved in forebrain development and regulation of neurogenesis.
The HMGB1, HMGB1L10, ID4, RUNX1 and
CBFA2T2 genes possessed the capabilities of transcription
factor binding, according to the molecular function terms (Table III).
| Table III.Functional annotation of the adjacent
genes with demethylated promoter regions in Down syndrome
samples. |
Table III.
Functional annotation of the adjacent
genes with demethylated promoter regions in Down syndrome
samples.
| Category | GO term | Genes (n) | Gene |
|---|
| BP | GO:0016481~negative
regulation of transcription | 5 | HMGB1, HMGB1L10,
ID4, LRRFIP1, CBFA2T2 |
| BP | GO:0002520~immune
system development | 3 | BARX1, POLM,
RUNX1 |
| BP |
GO:0032147~activation of protein kinase
activity | 2 | MAP3K4, STRADB |
| BP |
GO:0030855~epithelial cell
differentiation | 2 | BARX1, CBFA2T2 |
| BP |
GO:0030900~forebrain development | 2 | SLC1A2, ID4 |
| BP |
GO:0050767~regulation of neurogenesis | 2 | ID4, TIMP2 |
| CC |
GO:0005694~chromosome | 4 | HMGB1, BOD1, BARX1,
HMGB1L10 |
| CC |
GO:0019898~extrinsic to membrane | 3 | OSBP2, PCSK6,
FARP1 |
| MF |
GO:0008134~transcription factor
binding | 5 | HMGB1, HMGB1L10,
ID4, RUNX1, CBFA2T2 |
| MF |
GO:0016564~transcription repressor
activity | 3 | ID4, LRRFIP1,
CBFA2T2 |
| MF |
GO:0030528~transcription regulator
activity | 6 | BARX1, ATAD2, ID4,
LRRFIP1, RUNX1, CBFA2T2 |
Functional enrichment analysis showed that
ID4 was not only involved in neuronal differentiation, but
also functioned in transcriptional suppression. The demethylation
of its promoter region led to the increased expression level of
ID4 (Table III).
Analysis of DEGs
Combined with the RNA-seq data, the present study
analyzed transcriptome differences between the DS samples and
normal samples, and identified a total of 584 DEGs, including
RUNX1, which were upregulated (Table IV).
| Table IV.Number of differentially expressed
genes in Down syndrome samples, compared with control samples. |
Table IV.
Number of differentially expressed
genes in Down syndrome samples, compared with control samples.
| Expression | Genes (n) | Transcription
factors (n) |
|---|
| Upregulated | 584 | 24 |
| Downregulated | 0 | 0 |
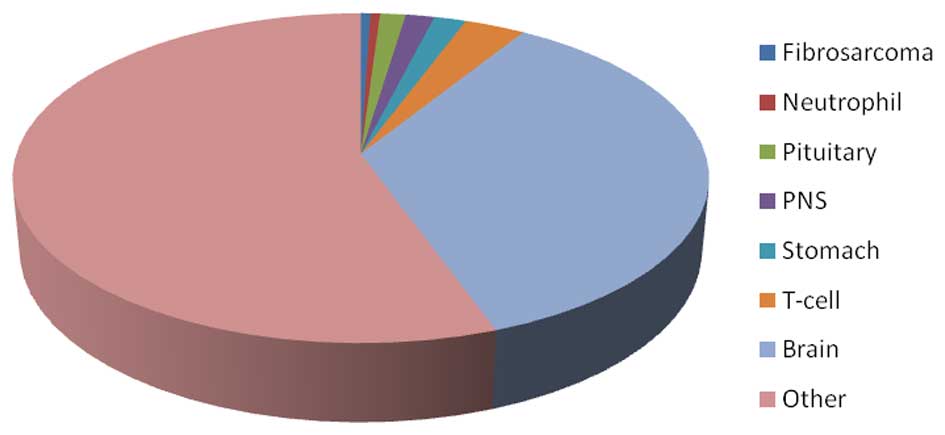
Based on the detection of tissue-specific gene
database, 208 of the 584 DEGs (36%) were found to have specific
expression in brain tissue. By contrast, 52 DEGs in the DS samples
were expressed specifically in neutrophils, the pituitary,
peripheral nervous system, stomach and T-cells, which was
substantially lower than the number of brain tissue-specific genes
(Fig. 2).
Finally, with the addition of transcription factor
data, the present study identified 24 DEGs with transcriptional
regulatory function, of which the eight transcription factors, zinc
finger protein 43, early growth response (EGR)3, nuclear
receptor subfamily 4, group A, member 2 (NR4A2), nuclear
receptor subfamily 3, group C, member 2 (NR3C2), LIM
homeobox 2, gastrulation brain homeobox 2, pentraxin-related gene,
rapidly induced by interleukin-1β and nuclear factor I/A, were
brain-tissue specific (Table
V).
| Table V.Differentially expressed genes with
transcriptional regulatory function and brain tissue
specificity. |
Table V.
Differentially expressed genes with
transcriptional regulatory function and brain tissue
specificity.
| Transcription
factor | Description | Brain-specific |
|---|
| ZNF43 | Zinc finger protein
43 | Yes |
| EGR3 | Early growth
response 3 | Yes |
| EGR2 | Early growth
response 2 | No |
| ZFY | Zinc finger
protein, Y-linked | No |
| MITF |
Microphthalmia-associated transcription
factor | No |
| NR4A2 | Nuclear receptor
subfamily 4, group A, member 2 | Yes |
| NR3C2 | Nuclear receptor
subfamily 3, group C, member 2 | Yes |
| FOXN1 | Forkhead box
N1 | No |
| HOXB13 | Homeobox B13 | No |
| VAX2 | Ventral anterior
homeobox 2 | No |
| HNF4G | Hepatocyte nuclear
factor 4, γ | No |
| SIX4 | SIX homeobox 4 | No |
| FOXP1 | Forkhead box
P1 | No |
| HESX1 | HESX homeobox
1 | No |
| ERCC8 | Excision repair
cross-complementing rodent repair deficiency, complementation group
8 | No |
| HOXA1 | Homeobox A1 | No |
| HOXB5 | Homeobox B5 | No |
| LHX2 | LIM homeobox 2 | Yes |
| PAX8 | Paired box 8 | No |
| GBX2 | Gastrulation brain
homeobox 2 | Yes |
| LHX6 | LIM homeobox 6 | No |
| PTX3 | Pentraxin-related
gene, rapidly induced by IL-1β | Yes |
| NFIA | Nuclear factor
I/A | Yes |
| NFIB | Nuclear factor
I/B | No |
Association between differential
methylation and dysregulation
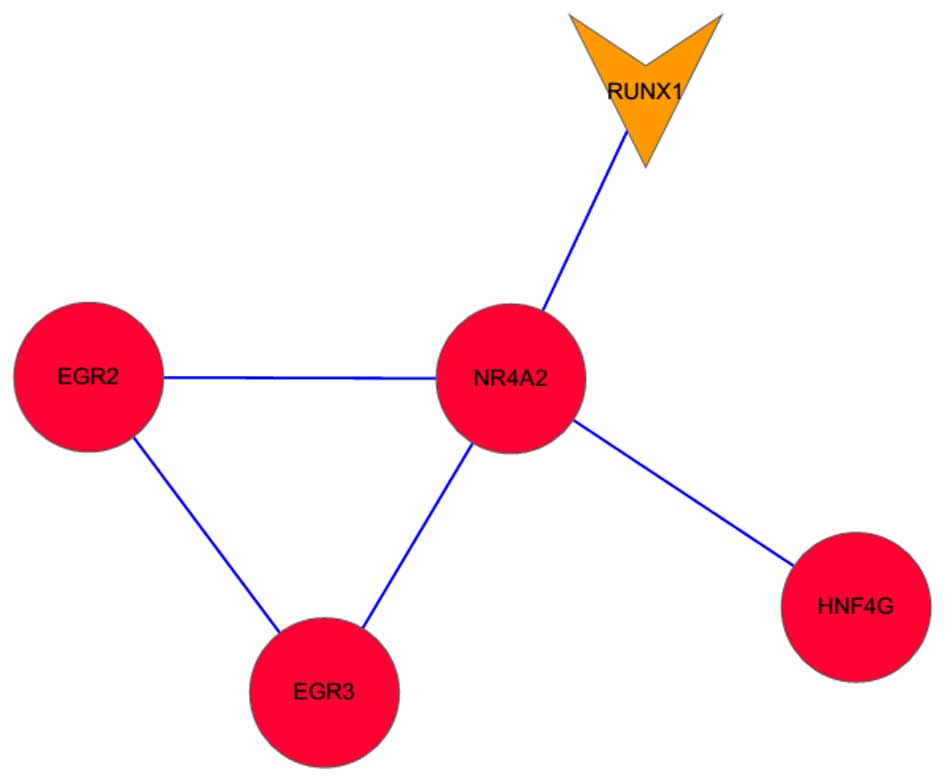
In order to examine the potential link between
abnormal methylation and dysregulation in DS samples, the present
study integrated their data combined with protein-protein
interactions the STRING database, and found only one PPI network
(Fig. 3). The network contained
five genes, including NR4A2, EGR2, EGR3, RUNX1 and hepatocyte
nuclear factor 4, γ (HNF4G). There were several interactions,
including RUNX1-NR4A2, NR4A2-EGR2 and NR4A2-EGR3, in the PPI
network.
Discussion
As a genetic disease in which an individual has 47
chromosomes instead of the usual 46 (25), DS affects ~1/730 live births and
occurs in all populations equally (26). In the present study, bioinformatics
tools were used to determine the genetic fragments associated with
DS. A total of 74 CpG regions had significant differential DNA
methylation between the DS and normal samples. There were five
abnormal DNA demethylated regions in chromosome 21. A total of 43
adjacent genes with demethylation in promoter regions and one
adjacent gene with upregulated methylation in promoter regions were
identified in the DS samples. In addition, 584 upregulated genes
were identified, including 24 genes with transcriptional regulatory
function. Only NR4A2, EGR2, EGR3, RUNX1 and HNF4G were involved in
the PPI network.
In the present study, upregulated RUNX1 was
located on chromosome 21, and the demethylation of the promoter
region of this gene was significant. Functional enrichment analysis
showed that RUNX1 was associated with immune system
development and possessed the capabilities of transcription factor
binding. It is reported that the expression of RUNX1 in
megakaryoblasts in children with DS and acute megakaryocytic
leukemia is lower, compared with cases of acute megakaryocytic
leukemia without DS (27,28). The risk of developing dementia of
Alzheimer's disease in individuals with DS is higher, compared with
that of the general population, and a variant within RUNX1
is closely linked with dementia of Alzheimer's disease in DS
(29). A previous study reported
that RUNX1 and NR4A2 can coordinately regulate the
differentiation of T cells (30,31).
The transcription factor, NURR1, which is also known as
NR4A2, is important in the functional maintenance,
development and survival of midbrain dopaminergic neurons (32). As with DS, Parkinson's disease is
also a disorder of the central nervous system, and decreased
expression levels of NURR1 may contribute to the identification of
Parkinson's disease and other neurlogical disorders (33). The transcription factors,
EGR2 and EGR3, are members of the Egr family, which
is involved in regulating the peripheral immune response, and
EGR2 may serve as a potential target in neuroinflammation
therapy for its host defense role in the central nervous system
immune response (34). According
to the results of the present study, transcription factors
EGR3 and NR4A2 were identified as brain-tissue
specific. In the PPI network, several interactions were identified,
including RUNX1-NR4A2, NR4A2-EGR2 and NR4A2-EGR3, indicating that
RUNX1 and NR4A2 may be involved in DS by coordinately
regulating EGR2 and EGR3.
As a member of the ID family, ID4 inhibits
the differentiation or the DNA binding of basic helix-loop-helix
transcription factors, regulating genes, which are important in
neuronal differentiation (35). A
previous study demonstrated that ID1, ID2, ID3
and ID4 are promising primary targets for methyl-CpG binding
protein 2-regulated neuronal maturation, which may be responsible
for the development of Rett syndrome, a neurodevelopmental disorder
(36). In the present study,
functional enrichment analysis indicated that ID4 possessed
the capabilities of transcription factor binding, and that
ID4 was involved in neuronal differentiation and
transcriptional suppression. Therefore, it was hypothesized that
the upregulated expression level of ID4 may be associated
with the symptoms of severe mental retardation and stunting of the
nervous system, which are observed in patients with DS.
In conclusion, the present study performed
integrated bioinformatics analyses of DNA methylation and RNA-seq
data to identify genes, which may be correlated with DS. A total of
43 adjacent genes with demethylation of promoter regions and one
adjacent gene with upregulated methylation of its promoter region
were identified in the DS samples. In addition, 584 upregulated
genes were identified, which included 24 genes with transcriptional
regulatory function. RUNX1, NR4A2, EGR2,
EGR3 and ID4 may be correlated with DS. However,
their mechanisms of action in DS remain to be fully elucidated and
further experimental validation is required.
References
|
1
|
Palomaki GE, Kloza EM, Lambert-Messerlian
GM, Haddow JE, Neveux LM, Ehrich M, van den Boom D, Bombard AT,
Deciu C, Grody WW, et al: DNA sequencing of maternal plasma to
detect Down syndrome: An international clinical validation study.
Genet Med. 13:913–920. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Palomaki GE, Deciu C, Kloza EM,
Lambert-Messerlian GM, Haddow JE, Neveux LM, Ehrich M, van den Boom
D, Bombard AT, Grody WW, et al: DNA sequencing of maternal plasma
reliably identifies trisomy 18 and trisomy 13 as well as Down
syndrome: An international collaborative study. Genet Med.
14:296–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khocht A, Yaskell T, Janal M, Turner BF,
Rams TE, Haffajee AD and Socransky SS: Subgingival microbiota in
adult Down syndrome periodontitis. J Periodontal Res. 47:500–507.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosdi M, Kadir A, Sheikh RS, Hj Murat Z
and Kamaruzaman N: The comparison of human body electromagnetic
radiation between down syndrome and non down syndrome person for
brain, chakra and energy field stability score analysis. 2012 IEEE
Control and System Graduate Research Colloquium. Malaysia. pp.
370–375. 2012;
|
|
5
|
Ward O: John Langdon Down: The man and the
message. Downs Syndr Res Pract. 6:19–24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cronk C, Crocker AC, Pueschel SM, Shea AM,
Zackai E, Pickens G and Reed RB: Growth charts for children with
Down syndrome: 1 month to 18 years of age. Pediatrics. 81:102–110.
1988.PubMed/NCBI
|
|
7
|
Myrelid A, Gustafsson J, Ollars B and
Annerén G: Growth charts for Down's syndrome from birth to 18 years
of age. Arch Dis Child. 87:97–103. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lott IT: Neurological phenotypes for Down
syndrome across the life span. Prog Brain Res. 197:1012012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roizen NJ and Patterson D: Down's
syndrome. Lancet. 361:1281–1289. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Freeman SB, Bean LH, Allen EG, Tinker SW,
Locke AE, Druschel C, Hobbs CA, Romitti PA, Royle MH, Torfs CP, et
al: Ethnicity, sex, and the incidence of congenital heart defects:
A report from the national down syndrome project. Genet Med.
10:173–180. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hernandez D and Fisher EM: Down syndrome
genetics: Unravelling a multifactorial disorder. Hum Mol Genet.
5:1411–1416. 1996.PubMed/NCBI
|
|
12
|
Patterson D and Costa AC: Down syndrome
and genetics-a case of linked histories. Nat Rev Genet. 6:137–147.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arron JR, Winslow MM, Polleri A, Chang CP,
Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, et al: NFAT
dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome
21. Nature. 441:595–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Altafaj X, Dierssen M, Baamonde C, Martí
E, Visa J, Guimerà J, Oset M, González JR, Flórez J, Fillat C and
Estivill X: Neurodevelopmental delay, motor abnormalities and
cognitive deficits in transgenic mice overexpressing Dyrk1A
(minibrain), a murine model of Down's syndrome. Hum Mol Genet.
10:1915–1923. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Voronov SV, Frere SG, Giovedi S, Pollina
EA, Borel C, Zhang H, Schmidt C, Akeson EC, Wenk MR, Cimasoni L, et
al: Synaptojanin 1-linked phosphoinositide dyshomeostasis and
cognitive deficits in mouse models of Down's syndrome. Proc Natl
Acad Sci USA. 105:9415–9420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng
DP, Holbrook JD, Law HY, Kwek KY, Yeo GS and Ding C: Global DNA
hypermethylation in down syndrome placenta. PLoS Genet.
9:e10035152013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krueger F and Andrews SR: Bismark: A
flexible aligner and methylation caller for Bisulfite-Seq
applications. Bioinformatics. 27:1571–1572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hebestreit K, Dugas M and Klein HU:
Detection of significantly differentially methylated regions in
targeted bisulfite sequencing data. Bioinformatics. 29:1647–1653.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mortazavi A, Williams BA, McCue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-Seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hsu H and Lachenbruch PA: Paired t test.
Wiley Encyclopedia of Clinical Trials. 1–3. 2008.
|
|
22
|
Huang DW, Sherman BT, Tan Q, Kir J, Liu D,
Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC and Lempicki RA:
DAVID bioinformatics resources: Expanded annotation database and
novel algorithms to better extract biology from large gene lists.
Nucleic Acids Res. 35:(Web Server issue). W169–W175. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jensen LJ, Kuhn M, Stark M, Chaffron S,
Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et
al: STRING 8-a global view on proteins and their functional
interactions in 630 organisms. Nucleic Acids Res. 37:(Database
issue). D412–D416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saito R, Smoot ME, Ono K, Ruscheinski J,
Wang PL, Lotia S, Pico AR, Bader GD and Ideker T: A travel guide to
Cytoscape plugins. Nat Methods. 9:1069–1076. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang Y, Mullaney KA, Peterhoff CM, Che S,
Schmidt SD, Boyer-Boiteau A, Ginsberg SD, Cataldo AM, Mathews PM
and Nixon RA: Alzheimer's-related endosome dysfunction in Down
syndrome is Abeta-independent but requires APP and is reversed by
BACE-1 inhibition. Proc Natl Acad Sci USA. 107:1630–1635. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van Gameren-Oosterom H, Fekkes M, van
Wouwe JP, Detmar SB, Oudesluys-Murphy AM and Verkerk PH: Problem
behavior of individuals with Down syndrome in a nationwide cohort
assessed in late adolescence. J Pediatr. 163:1396–1401. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bourquin JP, Subramanian A, Langebrake C,
Reinhardt D, Bernard O, Ballerini P, Baruchel A, Cavé H, Dastugue
N, Hasle H, et al: Identification of distinct molecular phenotypes
in acute megakaryoblastic leukemia by gene expression profiling.
Proc Natl Acad Sci USA. 103:3339–3344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Edwards H, Xie C, LaFiura KM, Dombkowski
AA, Buck SA, Boerner JL, Taub JW, Matherly LH and Ge Y: RUNX1
regulates phosphoinositide 3-kinase/AKT pathway: Role in
chemotherapy sensitivity in acute megakaryocytic leukemia. Blood.
114:2744–2752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Patel A, Rees SD, Kelly MA, Bain SC,
Barnett AH, Thalitaya D and Prasher VP: Association of variants
within APOE, SORL1, RUNX1, BACE1 and ALDH18A1 with dementia in
Alzheimer's disease in subjects with Down syndrome. Neurosci Lett.
487:144–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sekiya T, Kashiwagi I, Inoue N, Morita R,
Hori S, Waldmann H, Rudensky AY, Ichinose H, Metzger D, Chambon P
and Yoshimura A: The nuclear orphan receptor Nr4a2 induces Foxp3
and regulates differentiation of CD4+ T cells. Nat Commun.
2:2692011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Okada M, Hibino S, Someya K and Yoshmura
A: Regulation of regulatory T cells: Epigenetics and plasticity.
Adv Immunol. 124:249–273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jankovic J, Chen S and Le WD: The role of
Nurr1 in the development of dopaminergic neurons and Parkinson's
disease. Prog Neurobiol. 77:128–138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Le W, Pan T, Huang M, Xu P, Xie W, Zhu W,
Zhang X, Deng H and Jankovic J: Decreased NURR1 gene expression in
patients with Parkinson's disease. J Neurol Sci. 273:29–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yan Y, Tan X, Wu X, Shao B, Wu X, Cao J,
Xu J, Jin W, Li L, Xu W, et al: Involvement of early growth
response-2 (Egr-2) in lipopolysaccharide-induced neuroinflammation.
J Mol Histol. 44:249–257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Umetani N, Mori T, Koyanagi K, Shinozaki
M, Kim J, Giuliano AE and Hoon DS: Aberrant hypermethylation of ID4
gene promoter region increases risk of lymph node metastasis in T1
breast cancer. Oncogene. 24:4721–4727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peddada S, Yasui DH and LaSalle JM:
Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are
neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum
Mol Genet. 15:2003–2014. 2006. View Article : Google Scholar : PubMed/NCBI
|