Introduction
Increasing evidence indicates that abnormal growth
of glomerular mesangial cells (MCs) is an early event in various
glomerular diseases (1,2). The stimulation of MC proliferation in
response to glomerular injury has been attributed to numerous
factors, including high glucose levels (3), renin-angiotensin-aldosterone (Aldo)
system (RAAS) overactivation (4)
and platelet-derived growth factor (5). Aldo, which is considered to be a
dependent risk factor for chronic kidney disease, has become a
focus of investigations into the development and progression of
these diseases (6,7). However, the molecular mechanism
underlying Aldo-induced MC proliferation remains to be fully
elucidated.
Macroautophagy (hereafter referred to as autophagy)
is an evolutionarily conserved catabolic mechanism that is
responsible for the maintenance of energy homeostasis. Proteins
encoded by autophagy-related genes (Atgs), including Atg5 and Atg7,
are critical for the regulation of autophagy (8). The C-terminal carboxyl group of Atg12
is activated by the E1 enzyme Atg7 with consumption of ATP to form
a thioester bond with its catalytic cysteine residue, and
eventually attaches to the amino group of the lysine residue in
Atg5 via an isopeptide bond (9).
This process is critical for the formation of the autophagosomal
membrane. During autophagy, there may be an increased conversion of
microtubule-associated protein 1A/1B-light chain 3 (LC3)-I to
LC3-II in mammalian cells. In addition, p62, a selective substrate
of autophagy, may serve as a marker of autophagy (10). Autophagy, which contributes to the
abnormal proliferation of cancer cells (11,12),
has also been implicated in the physiological and
pathophysiological processes of numerous kidney diseases, including
diabetic nephropathy (13),
immunoglobulin A nephropathy (14)
and aging kidney (15,16).
The aim of the present study was to determine
whether Aldo induced MC autophagy, and if so, to further examine
the role of autophagy in Aldo-induced MC proliferation.
Materials and methods
Materials
The HBZY-1 rat MC line was obtained from the Chinese
Center for Type Culture Collection (Wuhan, China). Chloroquine
(CQ), 3-methyladenine (3-MA) and Aldo were purchased from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Mouse
anti-β-actin (catalog no. 3700), rabbit anti-Atg5 (catalog no.
12994), rabbit anti-Atg7 (catalog no. 8558), rabbit anti-LC3
(catalog no. 3868) and rabbit anti-p62 (catalog no. 5114)
antibodies were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). All other chemicals were of analytical
grade.
Cell culture and treatment
Cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal calf serum (FCS; Sigma-Aldrich;
Merck Millipore), 2 mM glutamine, 100 U/ml penicillin and 100 mg/ml
streptomycin, at 37°C in an atmosphere containing 5%
CO2. Following preincubation in serum-free DMEM for 24
h, cells were treated with 10−7 M Aldo for 24 h in DMEM
containing 10% FCS in the presence or absence of 10 µM CQ or 2 mM
3-MA. Control group cells treated with an identical volume of
PBS.
Cell proliferation assay
The [3H]thymidine assay was used as a
qualitative index of DNA synthesis, as previously reported
(17). Briefly, MCs were plated in
24-well plates and grown to 80% confluence. Cells were stimulated
with 10−7 M Aldo for 24 h. Subsequently, 1 µCi/ml
[3H]thymidine (PerkinElmer, Inc., Waltham, MA, USA) was
added to each well for 3 h. [3H]thymidine incorporation
into trichloroacetic acid-insoluble material was then measured
using a liquid scintillation spectrophotometer. To assess cell
number, MCs were stimulated in 6-well plates with 10−7 M
Aldo for 24 h and counted using a Z1-Coulter Counter (Beckman
Coulter, Inc., Brea, CA, USA).
Transient transfection of cells with
small interfering (si) RNA
For knockdown experiments, MCs were transiently
transfected with siRNA specifically targeting Atg7 or a negative
control siRNA using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. Cells were transfected with 20 nM Atg7 siRNA (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) or control siRNA (Santa Cruz
Biotechnology, Inc.) for 24 h prior to further treatment. Cellular
protein was extracted and subjected to Atg7 western blot
analysis.
Western blotting
Total cellular proteins were extracted by lysing
cells with buffer containing 150 mM NaCl, 0.1% Triton X-100, 0.5%
deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl (pH
7.0) and 1 mM ethylenediaminetetraacetic acid. Protein
concentrations were determined using the bicinchoninic acid method
(Beyotime Institute of Biotechnology, Haimen, China). MC protein
extracts (50 µg/lane) were separated by 10% SDS-polyacrylamide gel
electrophoresis and were transferred to nitrocellulose membranes.
Following blocking with 5% skimmed milk in Tris-buffered saline (pH
7.6) at room temperature for 1 h, the membranes were incubated at
4°C overnight with primary antibodies against LC3 (1:1,000), p62
(1:1,000), Atg7 (1:1,000) and β-actin (1:100). Membranes were then
incubated with Alexa Fluor 790-conjugated goat anti-rabbit
(1:2,500; catalog no. A27041; Thermo Fisher Scientific, Inc.) or
Alexa Fluor 680-conjugated goat anti-mouse (1:5,000; catalog no.
A28183; Thermo Fisher Scientific, Inc.) secondary antibodies, at
room temperature for 1 h. Protein bands were visualized with the
LI-COR Odyssey® protein analysis system (LI-COR
Biosciences, Lincoln, NE, USA). The relative intensity of each band
was normalized to β-actin using Image J software version 1.48
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Quantitative data are presented as the mean ±
standard deviation. Statistical significance was determined using
the unpaired Student's t-test or one-way analysis of variance
followed by Tukey's post-hoc test. SPSS software version 13.0
(SPSS, Inc., Chicago, IL, USA) was used for statistical analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
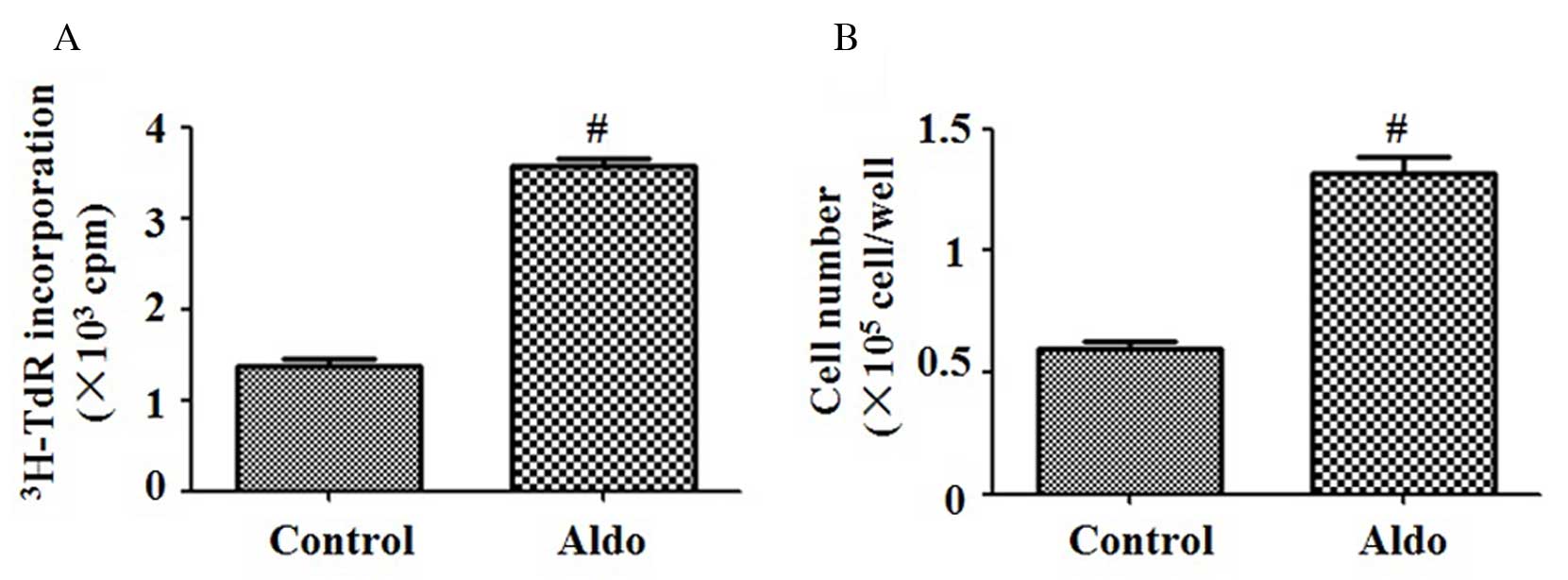
Aldo induces MC proliferation
[3H]thymidine incorporation and cell
counting assays were performed to evaluate MC proliferation.
Compared with the control group, 10−7 M Aldo
significantly increased the proliferation of MCs (P<0.0001;
Fig. 1A and B).
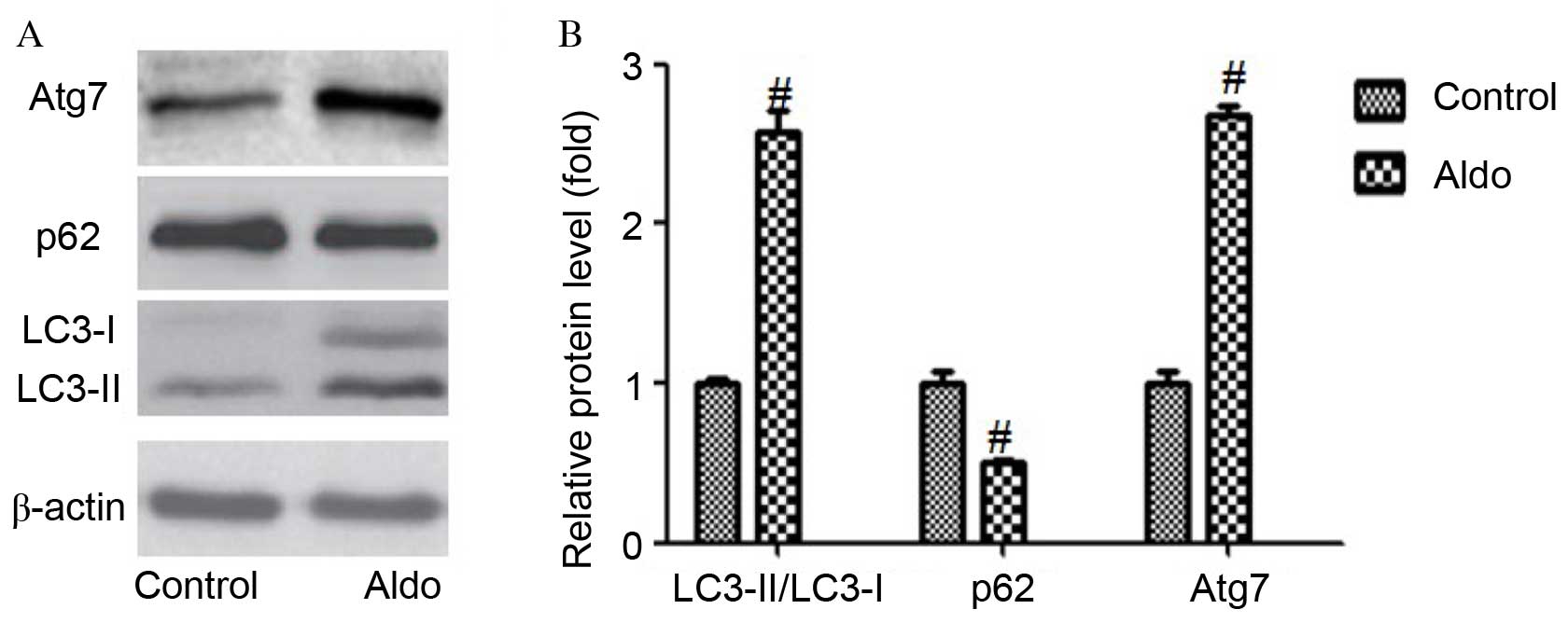
Aldo induces MC autophagy
As presented in Fig.
2, incubation with Aldo (10–7 M) for 24 h resulted in an
increased conversion from LC3-I to LC3-II (P=0.0003) and increased
expression levels of Atg7 (P<0.0001) compared with the control
group, as determined by western blotting. Furthermore, Aldo
increased the degradation of a selective substrate of autophagy,
p62 (P=0.0017). However, Aldo had no significant effect on the
protein expression levels of Atg5 (data not shown). These results
suggest that Aldo treatment promoted cellular autophagy in MCs.
Pharmacological inhibition of
autophagy prevents Aldo-induced MC proliferation
Numerous studies (18–20)
have described the use of pharmacological agents to demonstrate the
involvement of autophagy in the regulation of cell proliferation.
3-MA, which inhibits autophagosomal cargo-sequestration and CQ,
which acts on lysosmal function were used to inhibit individual
steps of the autophagic pathway. As presented in Fig. 3A and B, 3-MA blocked the conversion
from LC3-I to LC3-II (P<0.0001). Conversely, CQ increased the
levels of LC3-II/LC3-I. The opposite effects of 3-MA and CQ on the
level of LC3-II/LC3-I may be due to the fact that these two
inhibitors block autophagy at different points (P<0.0001). In
addition, 3-MA and CQ attenuated Aldo-induced MC proliferation
(P<0.0001; Fig. 3C).
siRNA-mediated knockdown of Atg7
inhibits Aldo-induced MC proliferation
To further support the finding that Aldo induced
proliferation via enhancing autophagy, cells were depleted of Atg7
using RNA interference technology, to investigate its impact on
Aldo-induced MC proliferation. Atg7 protein expression levels were
assessed by western blot analysis. Atg7 (P<0.0001) and
LC3-II/LC-I (P<0.0001) protein levels were markedly reduced in
MCs transfected with siRNA-Atg7 compared with those of control MCs
and MCs transfected with control siRNA (Fig. 4A and B). Furthermore, Atg7
silencing partially attenuated Aldo-induced MC proliferation
(P<0.0001; Fig. 4C).
Discussion
As a critical mediator of the RAAS, plasma and
tissue Aldo levels are elevated in patients with hypertension
(21), and diabetic and other
progressive nephropathies (22,23),
and may exert various physiological effects (24). Although interruption of the RAAS
with angiotensin-converting enzyme inhibitors and angiotensin
receptor blockers significantly improves kidney injury, Aldo
concentrations ‘escape’ to baseline during chronic therapy
(25). Increasing attention has
recently been focused on the role of Aldo in the development and
progression of kidney injury.
Elucidating the mechanisms underlying the induction
of MC proliferation by Aldo may contribute to the development of
novel and effective treatment strategies for glomerular diseases.
Huang et al (17)
demonstrated that Aldo stimulated MC proliferation via the
phosphoinositide 3-kinase/protein kinase B signaling pathway and
reactive oxygen species-dependent epithelial growth factor receptor
intracellular signaling. However, other, as yet unidentified,
mechanisms may contribute to this process.
Autophagy is a self-protection mechanism that is
triggered in response to harmful stimuli. A primary function of
autophagy is to transport the surrounding materials and damaged
organelles to lysosomes for degradation, thus enhancing
intracellular recycling of degraded metabolites to maintain the
energy metabolism cycle of cells. Autophagy fuels cancer cell
metabolism (9), and contributes to
abnormal proliferation (26),
invasion (27), and resistance to
chemotherapy and radiation therapy (28). In the present study, Aldo induced
autophagy as indicated by the increased expression levels of Atg7,
the increased conversion from LC3-I to LC3-II and the increased
degradation of p62, which is accompanied by MC proliferation.
Notably, pharmacological inhibition of autophagy or siRNA-mediated
knockdown of Atg7 attenuated Aldo-induced MC proliferation, thus
indicating that autophagy was at least partially responsible for
this effect.
In conclusion, the results of the present study may
partly explain the mechanism underlying the induction of abnormal
MC proliferation by Aldo. The results suggested that autophagy may
be a potential therapeutic target for the treatment of experimental
mesangial proliferative renal disease. As the present study was
performed in vitro, further in vivo studies are
required to determine whether autophagy is involved in Aldo-induced
MC proliferation in the kidney.
Acknowledgements
The present study was supported by a grant from the
Basic Research Projects of Science and Technology Bureau of
Changzhou City (grant no. CJ20140024).
References
|
1
|
Kurogi Y: Mesangial cell proliferation
inhibitors for the treatment of proliferative glomerular disease.
Med Res Rev. 23:15–31. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Feng Q, Huang S, Zhang A, Chen Q, Guo X,
Chen R and Yang T: Y-box protein 1 stimulates mesangial cell
proliferation via activation of ERK1/2. Nephron Exp Nephrol.
113:e16–e25. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu X, Fan Q, Xu L, Li L, Yue Y, Xu Y, Su
Y, Zhang D and Wang L: Ursolic acid attenuates diabetic mesangial
cell injury through the up-regulation of autophagy via
miRNA-21/PTEN/Akt/mTOR suppression. PLoS One. 10:e1174002015.
|
|
4
|
Yuan Y, Zhang A, Huang S, Ding G and Chen
R: A PPARgamma agonist inhibits aldosterone-induced mesangial cell
proliferation by blocking ROS-dependent EGFR intracellular
signaling. Am J Physiol Renal Physiol. 300:F393–F402. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang B, Zhang A, Zheng J, Gong J, Li S,
Zeng Z and Gan W: Bufalin inhibits platelet-derived growth
factor-BB-induced mesangial cell proliferation through mediating
cell cycle progression. Biol Pharm Bull. 34:967–973. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ajani AE, Marwick TH and Krum H:
Aldosterone antagonists: A new treatment option for patients with
post-myocardial infarction heart failure. Cardiovasc Revasc Med.
7:234–236. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fiebeler A and Haller H: Participation of
the mineralocorticoid receptor in cardiac and vascular remodeling.
Nephron Physiol. 94:p47–p50. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mizushima N, Noda T, Yoshimori T, Tanaka
Y, Ishii T, George MD, Klionsky DJ, Ohsumi M and Ohsumi Y: A
protein conjugation system essential for autophagy. Nature.
395:395–398. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang B, Ding W, Zhang M, Li H, Guo H, Lin
L, Chen J and Gu Y: Role of FOXO1 in aldosterone-induced autophagy:
A compensatory protective mechanism related to podocyte injury.
Oncotarget. 26–May;2016.(Epub ahead of print).
|
|
11
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu YL, Jahangiri A, Delay M and Aghi MK:
Tumor cell autophagy as an adaptive response mediating resistance
to treatments such as antiangiogenic therapy. Cancer Res.
72:4294–4299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiao T, Guan X, Nie L, Wang S, Sun L, He
T, Huang Y, Zhang J, Yang K, Wang J and Zhao J: Rapamycin promotes
podocyte autophagy and ameliorates renal injury in diabetic mice.
Mol Cell Biochem. 394:145–154. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sato S, Yanagihara T, Ghazizadeh M,
Ishizaki M, Adachi A, Sasaki Y, Igarashi T and Fukunaga Y:
Correlation of autophagy type in podocytes with histopathological
diagnosis of IgA nephropathy. Pathobiology. 76:221–226. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hartleben B, Gödel M, Meyer-Schwesinger C,
Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ,
Lindenmeyer MT, et al: Autophagy influences glomerular disease
susceptibility and maintains podocyte homeostasis in aging mice. J
Clin Invest. 120:1084–1096. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kume S, Uzu T, Horiike K, Chin-Kanasaki M,
Isshiki K, Araki S, Sugimoto T, Haneda M, Kashiwagi A and Koya D:
Calorie restriction enhances cell adaptation to hypoxia through
Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J
Clin Invest. 120:1043–1055. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang S, Zhang A, Ding G and Chen R:
Aldosterone-induced mesangial cell proliferation is mediated by EGF
receptor transactivation. Am J Physiol Renal Physiol.
296:F1323–F1333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Chen Z, Neelapu SS, Romaguera J
and McCarty N: Hedgehog inhibitors selectively target cell
migration and adhesion of mantle cell lymphoma in bone marrow
microenvironment. Oncotarget. 7:14350–14365. 2016.PubMed/NCBI
|
|
19
|
Button RW, Vincent JH, Strang CJ and Luo
S: Dual PI-3 kinase/mTOR inhibition impairs autophagy flux and
induces cell death independent of apoptosis and necroptosis.
Oncotarget. 7:5157–5175. 2016.PubMed/NCBI
|
|
20
|
Kowalik MA, Perra A, Ledda-Columbano GM,
Ippolito G, Piacentini M, Columbano A and Falasca L: Induction of
autophagy promotes the growth of early preneoplastic rat liver
nodules. Oncotarget. 7:5788–5799. 2016.PubMed/NCBI
|
|
21
|
Jugdutt BI: Expanding saga of the
Renin-Angiotensin system: The angiotensin II Counter-Regulatory AT2
receptor pathway. Circulation. 131:1380–1383. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mavrakanas TA, Gariani K and Martin PY:
Mineralocorticoid receptor blockade in addition to angiotensin
converting enzyme inhibitor or angiotensin II receptor blocker
treatment: An emerging paradigm in diabetic nephropathy: A
systematic review. Eur J Intern Med. 25:173–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rüster C and Wolf G:
Renin-angiotensin-aldosterone system and progression of renal
disease. J Am Soc Nephrol. 17:2985–2991. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boldyreff B and Wehling M: Non-genomic
actions of aldosterone: Mechanisms and consequences in kidney
cells. Nephrol Dial Transplant. 18:1693–1695. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schrier RW, Masoumi A and Elhassan E:
Aldosterone: Role in edematous disorders, hypertension, chronic
renal failure, and metabolic syndrome. Clin J Am Soc Nephrol.
5:1132–1140. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee WY, Hsu KF, Chiang TA and Chen CJ:
Phellinus linteus extract induces autophagy and synergizes with
5-fluorouracil to inhibit breast cancer cell growth. Nutr Cancer.
67:275–284. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang W, Li Q, Song C and Lao L: Knockdown
of autophagy-related protein 6, Beclin-1, decreases cell growth,
invasion, and metastasis and has a positive effect on
chemotherapy-induced cytotoxicity in osteosarcoma cells. Tumour
Biol. 36:2531–2539. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pennati M, Lopergolo A, Profumo V, De
Cesare M, Sbarra S, Valdagni R, Zaffaroni N, Gandellini P and
Folini M: MiR-205 impairs the autophagic flux and enhances
cisplatin cytotoxicity in castration-resistant prostate cancer
cells. Biochem Pharmacol. 87:579–597. 2014. View Article : Google Scholar : PubMed/NCBI
|