Introduction
Hereditary multiple exostoses (HME), also known as
multiple osteochondroma (MO), is an autosomal dominant bone
disorder with an incidence of 1 in 50,000 live births in western
populations (1). The morbidity
rate is greater in males than females, with a ratio of 1.5:1, due
to the mutations exerting a weaker phenotypic effect in females
(2). HME is characterized by the
presence of multiple benign cartilage-capped tumors, localized
primarily in the long tubular bones, particularly in the humerus
(10–50%), forearm (39–60%), knee (33%) and ankle (25%). The
majority of patients with HME (~70%) have a family history of the
condition (1,3,4).
HME is a genetically heterogeneous disorder with two
disease-causing genes identified, exostosin glycosyl transferase-1
(EXT1) and exostosin glycosyl transferase-2 (EXT2)
located at chromosomes 8q24 and 11p11-p12, respectively (5,6). The
proteins encoded by human EXT1 and EXT2 are type II
transmembrane glycoproteins, localized in the endoplasmic
reticulum. The EXT1/EXT2 complex is involved in the biosynthesis of
heparin sulphate (HS) proteoglycan (HSPG) (7). Prior to deacetylation, the EXT1/EXT2
complex catalyzes the elongation of the HS chain. EXT1 and EXT2 are
ubiquitously expressed in developing limb buds, and in
osteochondromas their expression was decreased in correlation with
mutation status (8,9). To date, ~422 separate pathogenic
mutations in EXT1 and ~221 mutations in EXT2 have
been identified. Mutations in EXT1 account for 56–78% of
cases in MO families, whereas EXT2 mutations have been
identified in 21–44% of cases (4,10–15).
However, in China, EXT2 mutations are identified more
frequently than EXT1 mutations (16). The majority of patients have been
identified in a single family or as sporadic cases. Nonsense,
frameshift and splice-site mutations, which represent the majority
of MO-causing mutations (80%), have been predicted to lead to the
premature translational termination of the associated amino acids,
and the subsequent production of a truncated protein (17). Mutations in EXT1 are
dispersed along the gene, and may occur in various exons (14,18);
however, EXT2 mutations do not appear to occur in the final
third of the gene-coding region (17).
Other than EXT1 and EXT2, the
EXT3 gene has been mapped to chromosome 19p (19). It appears to be a minor locus in
HME families and no causative mutations in EXT3 have been
identified (20). Three additional
EXT-like genes, designated EXTL1, EXTL2 and
EXTL3 have been identified and mapped to chromosomes 1
(1p36, 1p11-p12) and 8 (8p12) (21–23).
Although the EXTL genes are considered strong candidate
genes for MO, to date no HME family has been associated with these
loci.
The present study investigated a rare large family
with MO, and identified a novel splice-site mutation in
EXT2.
Materials and methods
Subjects
Written consent was obtained from all study
participants, and the present study was approved by the ethics
committee of The Second Xiangya Hospital (Changsha, China). The
proband was admitted to The Second Xiangya Hospital in 2012, and
presented with a large osteochondroma. The five-generation Chinese
family of the proband was subsequently investigated and a pedigree
constructed based on clinical and radiographical evaluations of all
family members (Fig. 1). Of the 33
family members, there were 13 affected individuals (11 males and 2
females) aged 8–80 years (average age, 52 years). The incidence
rate was therefore 40% within the family, and MO occurred in each
generation. The affected individuals had 6–16 exostoses, typically
located at the juxtaepiphyseal regions of long bones; however,
these were not as large as those present in the proband. There were
no other lesions that were atypical of MO and no evidence of short
stature (average height of adult male and female was 165 and 156
cm, respectively). All affected individuals had lesions detected
prior to age 10, but had never received surgery, with the exception
of the proband.
The proband, family member IV-2, a 42-year-old male,
presented with 41-year history of multiple osteochondroma. Physical
examination revealed a large mass on the left side of the back
(Fig. 2A), (39×33×19 cm), and
>14 osseous nodules of varying sizes located on the prothorax
wall, left scapula, bilateral forearms, knees and left ankle. The
patient had had an operation at age 22 due to a rapidly enlarging
lumbar spinal osteochondroma.
X-ray analysis revealed a diffuse flocculent shadow
with high density in the left lung field and multiple bony
protrusions on limbs, as presented in Fig. 2B. Computed tomography (CT)
angiography did not reveal any imaging of large arteries,
indicating a potential chondroma (Fig.
2C). CT revealed the left back mass was of mixed density and
multiple flecked calcifications, which spread into the left side of
the chest (Fig. 2D).
Histopathological analysis revealed an osteochondroma with focal
malignant transformation, as presented in Fig. 2E. Surgery was conducted at The
Second Xiangya hospital to remove the osteochondroma. The patient
was subsequently followed up once every three months in the first
year, and once every year thereafter. No recurrence has been
detected as of October 2015.
DNA analysis
DNA of all affected family members was extracted
from peripheral blood as previously described (24). The coding regions of the
EXT1 and EXT2 genes were amplified by polymerase
chain reaction (PCR) using primer sets, as previously described
(25). The PCR products were
purified by BigDye® Terminator version 1.1 (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
followed by direct DNA sequencing with an ABI 3100 Automatic
Sequencer (Applied Biosystems; Thermo Fisher Scientific, Inc.),
using forward and reverse primers.
Reverse transcription-PCR
(RT-PCR)
Total RNA was extracted from the tumor tissue of the
proband and healthy lung tissue of an unrelated individual, using
the QIAgen RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA),
according to the manufacturer's protocol. cDNA synthesis was
performed using RevertAid™ First Strand cDNA Synthesis kit
(Fermentas; Thermo Fisher Scientific, Inc.), using the following
primers: Forward, 5′-AACCAGAACACACTGCGCATCAAG and reverse,
5′-AGCTCCACGAAGAACCACACAGAA for exons 2–5. Amplification of cDNA
was performed and products were purified by BigDye Terminator
version 1.1 followed by direct DNA sequencing with an ABI 3100
Automatic Sequencer.
Results
DNA was extracted from family members with HME
(Fig. 1A, solid squares), and the
possible mutations residing in the EXT genes were scanned in
the exon and intron junctions. DNA sequence analysis revealed a
heterozygous mutation, c.939+1 G>T in EXT2 (Fig. 3) in all family members with HME;
none of the unaffected family members carried this mutation. This
indicated the association of this inheritable mutation with HME. No
other mutations were reported in any of the analyzed samples.
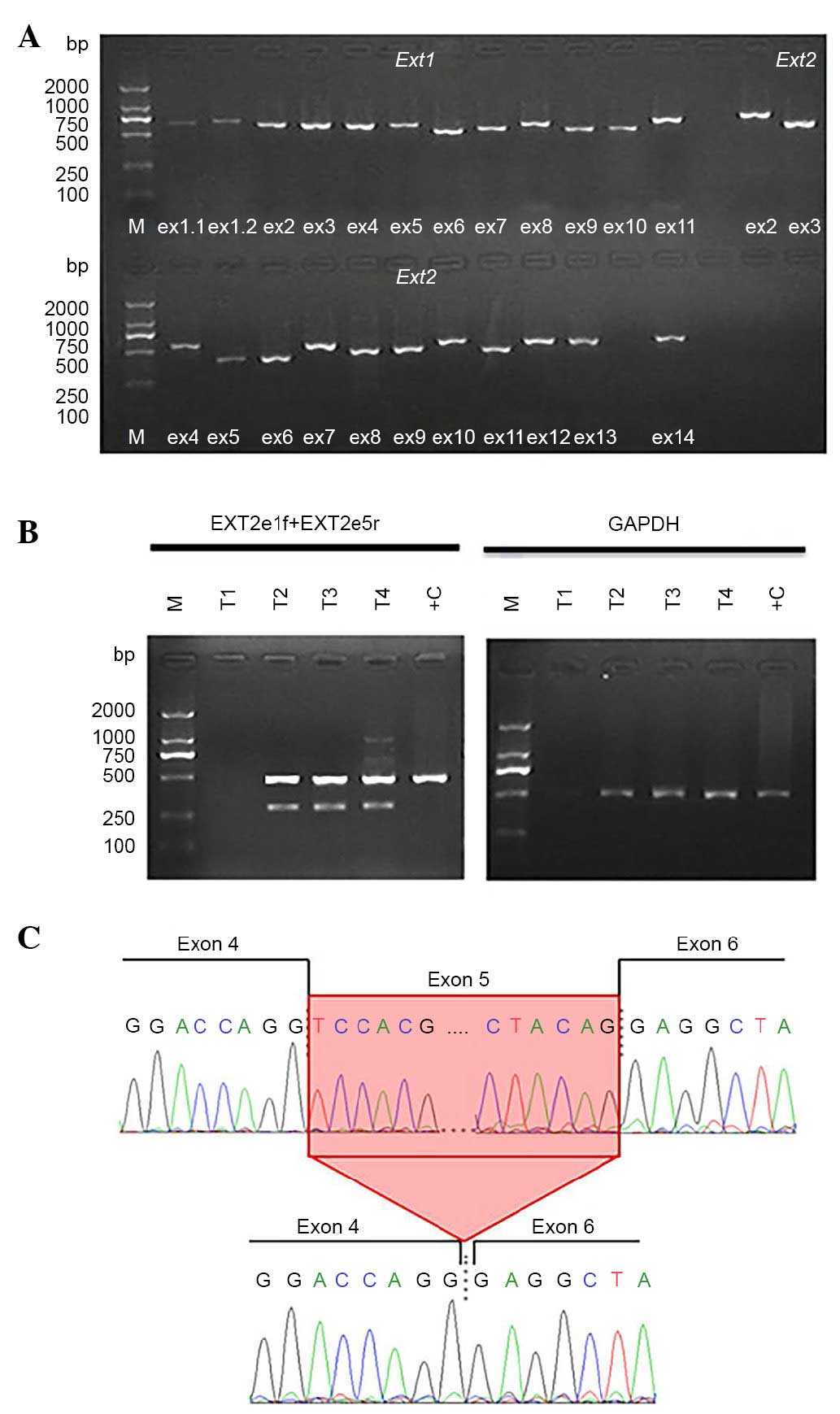
The result of PCR gel electrophoresis is presented
in Fig. 4. Following RT-PCR at
exons 2–5, two bands were identified in the proband; one band of
normal size (~500 bp) and one smaller band (~300 bp). The PCR
products were purified and sequenced. The size of one band was
revealed to be 429 bp, and the other, 296 bp. Compared with cDNA of
EXT2, sequence analysis confirmed exon 5 skipping in the
aberrant allele, resulting in an in-frame deletion of the
EXT2 protein.
Discussion
The MO family investigated in the present study
exhibited a ratio of male and female patients at 5.5:1, which
differs from the ratio of 1.5:1 reported previously (26). Therefore, the present study
performed a detailed physical examination of all healthy females in
the 3rd to 5th generation, to rule out misdiagnosis due to a weak
phenotype. It was concluded that the difference may be due to a
unique family structure.
Linkage analysis has confirmed that HME is
genetically heterogeneous, and the genes that have indicated the
greatest levels of association are EXT1, EXT2 and
EXT3. It has been revealed that mutations in EXT1 or
EXT2 are responsible for the majority of HME cases. The
proteins EXT1 and EXT2 form a hetero-oligomeric complex that
functions in HSPG biosynthesis. This complex has a substantially
greater glycosyltransferase activity than homo-oligomers of EXT1 or
EXT2 (27); therefore, a mutation
present in EXT1 or EXT2 may result in a critical
reduction in HSPG (9,28,29).
This may subsequently alter the balance of fibroblast growth factor
and Indian hedgehog homolog signals (30–32).
Thus, the normal signaling pathway involved in bone development may
be affected, leading to premature differentiation of cartilage,
cartilage cell proliferation and abnormal bone growth in the
adjacent areas (33), resulting in
HME.
According to the MO Mutation Database
(medgen.ua.ac.be/LOVDv.2.0/home.php), 713 mutations of the
EXT1 gene and 386 mutations of the EXT2 gene have
been identified; however, no mutations in EXT3 have been
reported. Among the 386 EXT2 mutations, the majority are
nonsense mutations, followed by frameshift and substitution
mutations. Mutations occur primarily in exons 2–8, seldom occurring
downstream. The present study revealed a novel splicing mutation
(C.939+1 G>T) leading to deletion of 196 bp in exon 5 of
EXT2, which may result in a truncated and subsequently
pathogenic protein. The EXT2 gene encodes a protein 718
amino acids in length. This mutation resulted in deletion from
codon 744 to 939 of exon 5 of the mRNA, causing a shift in the
codon-reading frame, followed by the synthesis of 266 novel amino
acids that terminate with a stop codon at position 994. Certain
studies have suggested that no mutations exist downstream of exon
8; however, according to the Human Gene Mutation Database
(www.hgmd.cf.ac.uk/ac/index.php), 3 patients with
mutations in exon 10 and 4 patients with mutations in exon 11 have
been identified. Therefore, mutations in the last 6 exons of
EXT2 are very rare. The truncated protein that arises due to
the C.939+1 G>T mutation in the family investigated in the
present study did not contain the amino acids encoded for by exon 7
to l4. This alteration may cause disease; however, whether it is
associated with the rare large osteochondroma that occurred on
proband remains to be elucidated. The present study suggested that
Knudson's two hit hypothesis or potential mutations in EXT3
or EXTL may explain the occurrence of the
osteochondroma.
In conclusion, the present study demonstrated that
the C.939+1 G>T (EXT2) mutation, present in a
five-generation 33-member MO family, resulted in the splicing out
of exon 5. These results have extended the mutational spectrum of
EXT2.
References
|
1
|
Schmale GA, Conrad EU III and Raskind WH:
The natural history of hereditary multiple exostoses. J Bone Joint
Surg Am. 76:986–992. 1994.PubMed/NCBI
|
|
2
|
Legeai-Mallet L, Munnich A, Maroteaux P
and Le Merrer M: Incomplete penetrance and expressivity skewing in
hereditary multiple exostoses. Clin Genet. 52:12–16. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saglik Y, Altay M, Unal VS, Basarir K and
Yildiz Y: Manifestations and management of osteochondromas: A
retrospective analysis of 382 patients. Acta Orthop Belg.
72:748–755. 2006.PubMed/NCBI
|
|
4
|
Porter DE, Lonie L, Fraser M, Dobson-Stone
C, Porter JR, Monaco AP and Simpson AH: Severity of disease and
risk of malignant change in hereditary multiple exostoses. A
genotype-phenotype study. J Bone Joint Surg Br. 86:1041–1046. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu YQ, Heutink P, de Vries BB, Sandkuijl
LA, van den Ouweland AM, Niermeijer MF, Galjaard H, Reyniers E,
Willems PJ and Halley DJ: Assignment of a second locus for multiple
exostoses to the pericentromeric region of chromosome 11. Hum Mol
Genet. 3:167–171. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wuyts W, Van Hul W, Wauters J, Nemtsova M,
Reyniers E, Van Hul EV, De Boulle K, de Vries BB, Hendrickx J,
Herrygers I, et al: Positional cloning of a gene involved in
hereditary multiple exostoses. Hum Mol Genet. 5:1547–1557. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esko JD and Selleck SB: Order out of
chaos: Assembly of ligand binding sites in heparan sulfate. Annu
Rev Biochem. 71:435–471. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lind T, Tufaro F, McCormick C, Lindahl U
and Lidholt K: The putative tumor suppressors EXT1 and EXT2 are
glycosyltransferases required for the biosynthesis of heparan
sulfate. J Biol Chem. 273:26265–26268. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hameetman L, David G, Yavas A, White SJ,
Taminiau AH, Cleton-Jansen AM, Hogendoorn PC and Bovée JV:
Decreased EXT expression and intracellular accumulation of heparan
sulphate proteoglycan in osteochondromas and peripheral
chondrosarcomas. J Pathol. 211:399–409. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hall CR, Cole WG, Haynes R and Hecht JT:
Reevaluation of a genetic model for the development of exostosis in
hereditary multiple exostosis. Am J Med Genet. 112:1–5. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Signori E, Massi E, Matera MG, Poscente M,
Gravina C, Falcone G, Rosa MA, Rinaldi M, Wuyts W, Seripa D, et al:
A combined analytical approach reveals novel EXT1/2 gene mutations
in a large cohort of Italian multiple osteochondromas patients.
Genes Chromosomes Cancer. 46:470–477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
White SJ, Vink GR, Kriek M, Wuyts W,
Schouten J, Bakker B, Breuning MH and den Dunnen JT: Two-color
multiplex ligation-dependent probe amplification: Detecting genomic
rearrangements in hereditary multiple exostoses. Hum Mutat.
24:86–92. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pedrini E, De Luca A, Valente EM, Maini V,
Capponcelli S, Mordenti M, Mingarelli R, Sangiorgi L and
Dallapiccola B: Novel EXT1 and EXT2 mutations identified by DHPLC
in Italian patients with multiple osteochondromas. Hum Mutat.
26:2802005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lonie L, Porter DE, Fraser M, Cole T, Wise
C, Yates L, Wakeling E, Blair E, Morava E, Monaco AP and Ragoussis
J: Determination of the mutation spectrum of the EXT1/EXT2 genes in
British Caucasian patients with multiple osteochondromas, and
exclusion of six candidate genes in EXT negative cases. Hum Mutat.
27:11602006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jennes I, Entius MM, Van Hul E, Parra A,
Sangiorgi L and Wuyts W: Mutation screening of EXT1 and EXT2 by
denaturing high-performance liquid chromatography, direct
sequencing analysis, fluorescence in situ hybridization, and a new
multiplex ligation-dependent probe amplification probe set in
patients with multiple osteochondromas. J Mol Diagn. 10:85–92.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu L, Xia J, Jiang H, Zhou J, Li H, Wang
D, Pan Q, Long Z, Fan C and Deng HX: Mutation analysis of
hereditary multiple exostoses in the Chinese. Hum Genet. 105:45–50.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wuyts W and Van Hul W: Molecular basis of
multiple exostoses: Mutations in the EXT1 and EXT2 genes. Hum
Mutat. 15:220–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raskind WH, Conrad EU III, Matsushita M,
Wijsman EM, Wells DE, Chapman N, Sandell LJ, Wagner M and Houck J:
Evaluation of locus heterogeneity and EXT1 mutations in 34 families
with hereditary multiple exostoses. Hum Mutat. 11:231–239. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Le Merrer M, Legeai-Mallet L, Jeannin P,
Horsthemke B, Schinzel A, Plauchu H, Toutain A, Achard F, Munnich A
and Maroteaux P: A gene for hereditary multiple exostosesmaps to
chromosome 19 p. Hum Mol Genet. 5:717–722. 1994. View Article : Google Scholar
|
|
20
|
Francannet C, Cohen-Tanugi A, Le Merrer M,
Munnich A, Bonaventure J and Legeai-Mallet L: Genotype-phenotype
correlation in hereditary multiple exostoses. J Med Genet.
7:430–434. 2001. View Article : Google Scholar
|
|
21
|
Wise CA, Clines GA, Massa H, Trask BJ and
Lovett M: Identification and localization of the gene for EXTL, a
third member of the multiple exostoses gene family. Genome Res.
7:10–16. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wuyts W, Van Hul W, Hendrickx J, Speleman
F, Wauters J, De Boulle K, Van Roy N, Van Agtmael T, Bossuyt P and
Willems PJ: Identification and characterization of a novel member
of the EXT gene family, EXTL2. Eur J Hum Genet. 5:382–389.
1997.PubMed/NCBI
|
|
23
|
Van Hul W, Wuyts W, Hendrickx J, Speleman
F, Wauters J, De Boulle K, Van Roy N, Bossuyt P and Willems PJ:
Identification of the third EXT-like gene (EXTL 3) belonging to the
EXT gene family. Genomics. 47:230–237. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miller SA and James RH: Variables
associated with ultraviolet transmittance measurements of
intraocular lenses. Am J Ophthalmol. 106:256–260. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wuyts W, Van Hul W, De Boulle K, Hendrickx
J, Bakker E, Vanhoenacker F, Mollica F, Lüdecke HJ, Sayli BS,
Pazzaglia UE, et al: Mutations in the EXT1 and EXT2 genes in
hereditary multiple exostoses. Am J Hum Genet. 62:346–354. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wicklund CL, Pauli RM, Johnston D and
Hecht JT: Natural history study of hereditary multiple exostoses.
Am J Med Genet. 55:43–46. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kitagawa H, Shimakawa H and Sugahara K:
The tumor suppressor EXT-like gene EXTL2 encodes an alpha1,
4-N-acetylhexosaminyltransferase that transfers
N-acetylgalactosamine and N-acetylglucosamine to the common
glycosaminoglycan-protein linkage region. The key enzyme for the
chain initiation of heparan sulfate. J Biol Chem. 274:13933–13937.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Romeo S, Oosting J, Rozeman LB, Hameetman
L, Taminiau AH, Cleton-Jansen AM, Bovée JV and Hogendoorn PC: The
role of noncartilage-specific molecules in differentiation of
cartilaginous tumors: Lessons from chondroblastoma and
chondromyxoid fibroma. Cancer. 110:385–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bornemann DJ, Duncan JE, Staatz W, Selleck
S and Warrior R: Abrogation of heparan sulfate synthesis in
drosophila disrupts the wingless, hedgehog and decapentaplegic
signaling pathways. Development. 131:1927–1938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bellaiche Y, The I and Perrimon N:
Tout-velu is a Drosophila homologue of the putative tumour
suppressor EXT-1 and is needed for Hh diffusion. Nature. 394:85–88.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lai LP and Mitchell J: Indian hedgehog:
Its roles and regulation in endochondral bone development. J Cell
Biochem. 96:1163–1173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Duncan G, McCormick C and Tufaro F: The
link between heparan sulfate and hereditary bone disease: Finding a
function for the EXT family of putative tumor suppressor proteins.
J Clin Invest. 108:511–516. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wuyts W, Radersma R, Storm K and Vits L:
An optimized DHPLC protocol for molecular testing of the EXT1 and
EXT2 genes in hereditary multiple osteochondromas. Clin Genet.
68:542–547. 2005. View Article : Google Scholar : PubMed/NCBI
|