Introduction
Dysregulation of the immune system leads to multiple
pathophysiological processes due to loss of the capacity to detect
and eliminate antigens by not distinguishing between self and
non-self (1). Generally, the
immune system can be divided into adaptive and innate immunity. It
is known that the complex processes of the adaptive immune system
enable B and T cells to detect non-self structures in B and T cells
through antigen-specific receptors. The adaptive immune system
exists only in vertebrates, whereas the innate immune system is
present in all multicellular organisms, indicating that it is an
evolutionarily conserved system (2). Sensing and excluding of invading
pathogens by innate immune cells is dependent on the molecular
structures that are shared by many pathogens but not expressed by
the host. These structures are detected through multiple-encoded
receptors or molecules, collectively named pattern recognition
receptors (PRRs) (3).
An important class of PRRs is the Toll-like receptor
(TLR) family that was identified recently in mammals and consists
of multiple members. TLRs were found during investigations
involving the search for proteins with sequence homology in the
intracellular domain of the interleukin (IL)-1 receptor and
Drosophila Toll molecules (4–6).
Drosophila Toll molecules originally were characterized as a
protein involved in the establishment of the dorsoventral polarity
in developing fly embryos, and then in the resistance of adult
flies to fungal infections. Since then, TLR genes have been
identified in the mouse and human genomes (7). The effects of TLR in microbial
product were first reported in 1998. Null mutations and missense
mutations of TLR4 gene lead to loss of the capacity in
response to lipopolysaccharide (LPS), which was the major component
of gram-negative bacteria (8,9).
TLRs have been shown to recognize a broad range of
microbial structures (10,11). Following activation, most TLRs
induce a common intracellular signaling pathway that culminates in
activation of the nuclear factor (NF)-κB, a key transcription
factor involved in activating the expression of chemokines,
cytokines, and cell-surface molecules such as adhesions, selectins,
integrins, and co-stimulatory molecules (10–12).
Initially, TLR4 was thought to be expressed solely in the immune
system. However, kidney mesangial and tubular cells also express
TLR4 (7,13).
Blood monocytes are an intermediate stage of
development that further differentiates tissues into various types
of macrophages populations. Tissue macrophages and other cells of
the innate immune system are critical for these surveillance and
defense activities (14).
Monocytes are activated by microbial components such as LPS and
12-O-tetradecanoylphorbol-13-acetate (TPA) that are recognized by
TLRs. After ligand binding, TLRs transduce extracellular signaling
to the nucleus and induce cytokine production and ultimately clear
the infection. Although currently 10 different human TLRs have been
characterized (10–12,15),
cytokine-mediated regulation of TLR expression remains poorly
understood. TLR expression analysis in primary human leukocytes
showed that professional phagocytes express the most varied TLR
profile, with CD14+ mononuclear cells expressing the
greatest amount of TLR-2, −4 and −8 (16,17).
In monocytes, the constant expression of TLR4 is important in
accepting bacterial infection signaling molecules such as LPS,
which functions as the main ligand binding to TLR4 to induce
inflammation. However, TLR4 signaling requires other binding
partners, including myeloid differentiation factor 88 (MyD88) as an
adaptor and NF-κB as a transcription factor. LPS binding occurs via
the coordinated sequence of binding events between soluble and cell
membrane proteins including LPS-binding protein, myeloid
differentiation-2 (MD-2) protein and CD14 (15). CD14 is a key LPS co-receptor,
pivotal in the initial binding of LPS and transfer of LPS to the
MD2/TLR4 complex to initiate signal signaling cascades (16).
In the present study, constant stimulation by high
concentration of LPS was used to determine whether LPS moderately
upregulated TLR4 expression through a positive feedback pathway in
a NF-κB-dependent manner. The results showed upregulated TLR4 at
the protein and mRNA levels enriched through the TLR4 modulation
style. However, cytokines such as tumor necrosis factor (TNF)-α and
IL-2 were not detected. Thus, the positive feedback mechanism
facilitates monocytes to differentiate into macrophages and rapidly
enhances multiple cytokine expression (known as ‘a cytokine storm’)
in order to react with the extracellular stress.
Materials and methods
Antibodies and reagents
CLI-095, also known as TAK-242, is a novel
cyclohexene derivative that suppresses TLR4 signaling specifically.
CLI-095 was purchased from InvivoGen (San Diego, CA, USA).
Helenalin, an NF-κB inhibitor, was purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA), and LPS (Escherichia
coli) and β-actin were purchased from Sigma-Aldrich (St. Louis,
MO, USA). The Alexa Fluor 488-labeled mouse anti-human antibodies
against TLR4 and Alexa Fluor 488-labeled isotype controls (mouse
IgG1) were obtained from BD Biosciences (Franklin Lakes, NJ, USA).
Monoclonal rabbit NF-κB antibody (cat. no. 8242; dilution,
1:1,000), monoclonal rabbit NF-κB pho-Ser468 antibody (cat. no.
2642; dilution, 1:1,000) and tyrosine phosphorylation antibody were
from Cell Signaling Technology, Inc. (Beverly, MA, USA). STAT3
inhibitor was STA-21 (sc-200757), 2 µmmol/l and NF-κB inhibitor was
helenalin (sc-218579) (both from Santa Cruz Biotechnology,
Inc.).
Cell culture
Human THP1 cells were treated in a time- and
concentration-dependent manner. The cells were divided into the low
LPS group, which was treated with 1 ng/ml LPS for 15 min; the high
LPS group, treated with 20 ng/ml LPS for 2 h; and the mock group,
treated with isovolumetric fetal bovine serum (FBS) for 15 min.
The cells were cultured with RPMI-1640 medium
supplemented with 20% heat-inactivated FBS, 2 mmol/l L-glutamine,
antibiotics (50 U/ml penicillin and 50 µg/ml streptomycin), and
maintained at 37°C in a humidified 5% CO2 atmosphere.
The differentiation of THP1 cells to macrophages involved a 6-day
protocol and 3-day exposure to LPS (5 nmol/l), followed by 3 days
of culture in the absence of LPS. Both concentrations of LPS were
used to induce optimal macrophage differentiation.
Determination of TLR4 by FACS
analysis
The levels of TLR4 were determined using FACScan
analysis. Cells (1×106) were re-suspended in
phosphate-buffered saline (PBS) containing 0.1% sodium azide and 5%
FBS was incubated in ice for 30 min, followed by incubation with
Alexa Fluor 488-conjugated anti-TLR4 antibody for 1 h. The cells
were washed twice, fixed in 2% formaldehyde in PBS and analyzed
using FACScan analysis. Negative controls were stained with
isotype-matched Alexa Fluor 488-conjugated IgG and compensation was
adjusted using the single-stained cell samples. Fluorescence
intensities were determined using FlowJo software (Tree Star Inc.,
Ashland, OR, USA).
Semi-quantitative and RT-PCR
Reverse transcription was performed in a 20-µl
reaction system with a total of 2 µg of RNA M-MLV reverse
transcriptase (Toyobo, Osaka, Japan). Quantitative RT-PCR and
RT-PCR were performed with an ABI PCR Thermal Cycler Dice detection
system and SYBR-Green dye (Toyobo) according to the manufacturer's
instructions. The primers used were: human TLR4 forward,
TGGGCAACCTGCTCTACCTA and reverse, GCTGTAGCTCGTTGGCAGA; and GAPDH
forward, ACAACTTTGGTATCGTGGAAGG and reverse,
GCCATCACGCCACAGTTTC.
Chromatin immunoprecipitation (ChIP)
and quantitative PCR
The method of treatment for THP1 cells is shown in
Fig. 2. The cells were cross
linked with 1% formaldehyde (Sigma-Aldrich) at 25°C for 15 min,
rinsed twice with ice-cold PBS (pH 7.4), and collected in PBS prior
to centrifugation at 2,000 × g for 5 min. Then cells were lysed in
1 ml of ChIP lysis buffer [50 mmol/l Tris-HCl (pH 8.1), 1% SDS and
10 mmol/l EDTA] and sonicated for 20 min with 30 sec on and off
cycles at high settings (Bioruptor; Diagenode, Denville, NJ, USA)
to produce chromatin fragments of 0.5 kb on average. Following
centrifugation and quantification, 50 µl of the supernatants were
used as inputs and the remainder was diluted 10-fold in IP buffer
[10 mmol/l Tris-HCl (pH 8.1), 1% Triton X-100, 0.1% deoxycholate,
0.1% SDS, 90 mmol/l NaCl and 2 mmol/l EDTA] and an equivalent of
2×106 cells/time-point was subjected to IP overnight at
4°C, followed by 2 h pre-clearing with 20 µl of a 50% protein
A/G-Sepharose beads (GE) slurry. These beads were prepared by two
washings in IP buffer and a 3-h incubation with 25 µg shared salmon
DNA and 200 µg BSA/ml of solution. The beads were then resuspended
1:1 in IP buffer and used for ChIPs. Anti-NF-κB (4 µl) and anti-RNA
polymerase II (4 µl) were used in each ChIP experiment. Complexes
were then recovered with 2-h incubation at 4°C with 20 µl of the
protein A/G-Sepharose beads solution. The precipitates were washed,
the beads removed and the DNA was precipitated by ethanol at −80°C
for >2 h and then centrifuged at 9,000 × g for 10 min. The
pellet was washed in 70% ethanol and recovered by TE buffer (pH
7.5).
Western blotting
Cells were collected and washed twice in PBS and the
cell lysates were obtained by adding RIPA lysis buffer for 20 min,
and centrifuging for 15 min at 9,000 × g. The supernatant was
obtained and densitometric analysis was performed with Quantity One
software (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The
relative expression level of each protein was normalized by
dividing the level of target protein by the level of GAPDH for each
sample.
Statistical analysis
Statistical analysis was performed using the
Statistical Package for Social Sciences (SPSS) for Windows (version
12.0; SPSS, Inc., Chicago, IL, USA). Student's t-tests were used to
determine the statistical significance of the differences between
the experimental groups. P<0.05 was considered to indicate a
statistically significant difference. The graphs were created using
Microcal Origin software (version 3.78; MicroCal Inc., Northampton,
MA, USA).
Results
LPS moderately upregulates TLR4
expression in THP1 cells
Monocytes are activated by microbial components that
are recognized by TLRs. After ligand binding, TLRs induce cytokine
production and ultimately clear the infection. However, except for
TLR activation, changes in the expression levels of TLRs following
ligand binding were detected with difficulty. It was previously
reported that the gene expression of TLR2 although not of TLR4 was
induced by LPS in mouse macrophages (7).
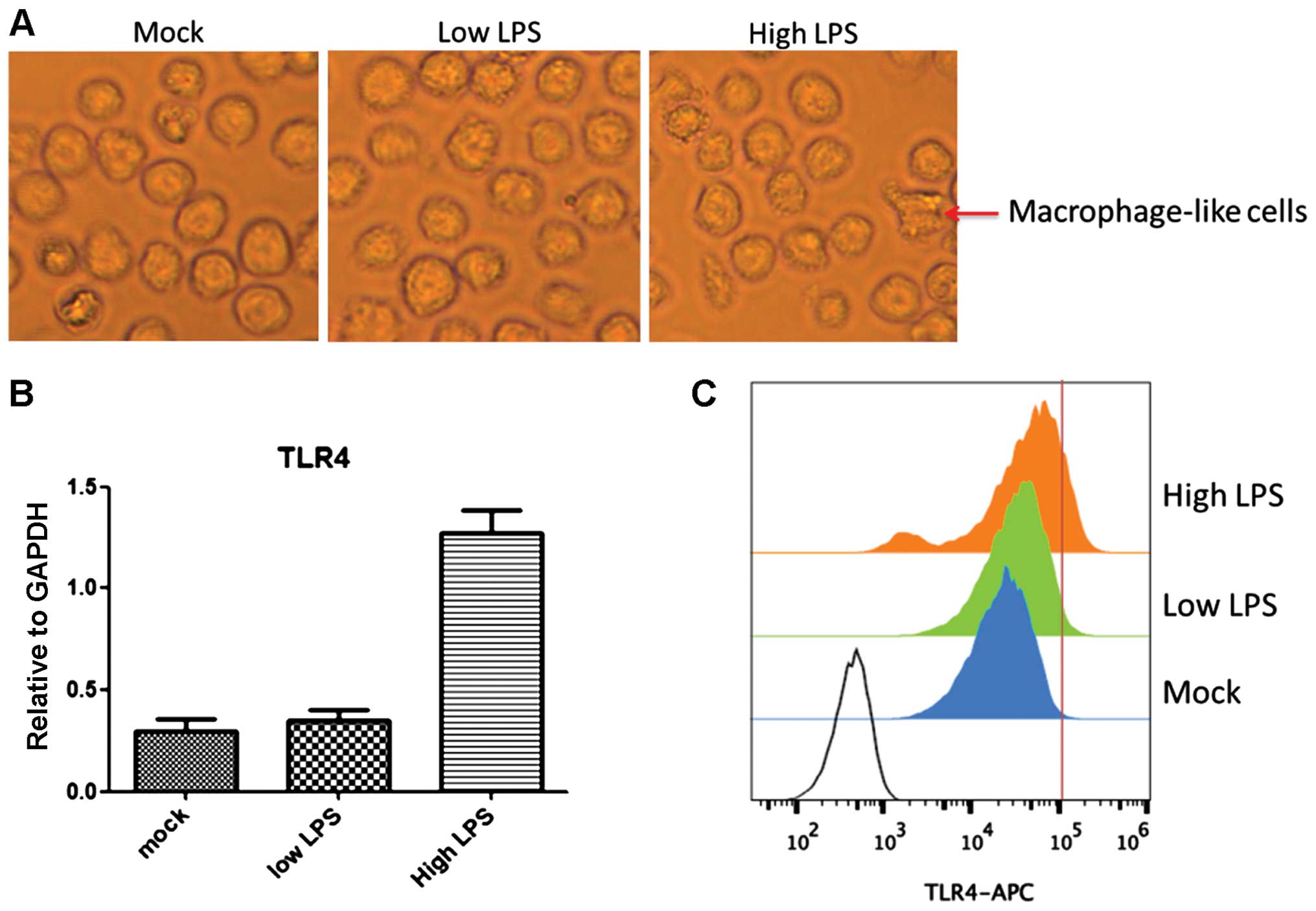
We found that TLR4 expression was upregulated by LPS
treatment in a time- and concentration-dependent manner.
Stimulation with 1 ng/ml LPS (low LPS) for 15 min had no obvious
effect on TLR4 expression enhancement. By contrast, stimuli with 20
ng/ml LPS (high LPS) for 2 h increased TLR4 expression 4-fold
compared to the level with low LPS (P<0.05) (Fig. 1B and C). Similarly, most THP1 cells
in the high LPS group differentiated to macrophage-like cells,
which did not occur in the low LPS group (Fig. 1A). A high concentration of LPS led
to microbial infection and the rapid upregulation of TLR4
facilitates clearance of these extracellular invaders. These
results suggested that the TLR4 transcriptional level was tightly
regulated by a feedback mechanism following extracellular
stimuli.
LPS-induced TLR4 activation is
required for upregulation of TLR4 expression
Activation of TLR4 by LPS only required 15 min, but
the prolongation of stimulation had an obvious effect on
upregulating TLR4 expression. The activation efficiency of TLR4 was
dependent on the increased expression in the cell membrane, and
continuous stimulation resulted in the monocytes obtaining the
capacity to respond to external stimuli because of ever-increasing
amounts of TLR4.
Compared to the mock and low LPS group, the tyrosine
phosphorylation of TLR4 tyrosine in the high LPS group presented
obvious enhancement. The downstream target of TLR4 signaling,
NF-κB, in the high LPS group was also strongly activated (Fig. 2A). When treated with the TLR4
inhibitor, the expression of TLR4 decreased and the TLR4 activation
was blocked (Fig. 2B and C).
Suspended THP1 cell differentiation from monocyte to macrophage was
also inhibited. The upregulation of TLR4 expression was increased
by TLR4 activation, whereas the expression of TLR4 was decreased
following stimulation by the TLR4 inhibitor. Thus, TLR4 activation
was required to upregulate TLR4 expression.
NF-κB targets TLR4 in chromatin and
promotes TLR4 expression
After TLR4 activation, MyD88 and other adaptors
triggered a cascade of cell signals through the intracellular
signal transduction domain such as NF-κB, signal transducer and
activator of transcription (STAT)-3 and −1, or STAT1 and STAT3,
thereby inducing the expression of inflammatory genes. To clarify
TLR4 promoter, we used ChIP to study the regulation of TLR4
expression. LPS stimulation resulted in NF-κB targeting TLR4
promoter while STAT3 STAT1 and STAT1 STAT3 had no obvious binding
to the TLR4 promoter (Fig. 3A),
indicating that TLR4 activation was transduced by LPS signaling to
NF-κB and not via STAT3 or STAT1 in THP1 cells. Furthermore, to
identify the effect of NF-κB inhibitor on TLR4 expression by
blocking the NF-κB transcription activity, we found that blocking
NF-κB transcription activity resulted in markedly and rapidly
decreased TLR4 expression. However, blocking NF-κB transcription
activity had no effect on TLR4 tyrosine phosphorylation. NF-κB
functioned as a transcription factor targeting chromatin, while
TLR4 was stimulated by LPS and then phosphorylated.
 | Figure 3.NF-κB targeted TLR4 in chromatin and
promotes TLR4 expression. (A) ChIP assay for NF-κB binding to TLR4
gene promoter. (B) Blocking NF-κB activation blocked TLR4
expression. (C) TLR4 protein expression in the cell membrane by
FACS. Three group treatment: i) Mock group, LPS stimulation without
any inhibitor; ii) STAT3 inhibitor group, LPS stimulation with
STAT3 inhibitor; and iii) NF-κB inhibitor group, LPS stimulation
with NF-κB inhibitor. LPS stimulation condition was 4 h and at 20
ng/ml LPS. STAT3 inhibitor was STA-21 (sc-200757; Santa Cruz
Biotechnology, Inc.), 2 µmmol/l. NF-κB inhibitor was helenalin
(sc-218579; Santa Cruz Biotechnology, Inc.), 5 µmmol/l. NF-κB,
nuclear factor-κB; TLR4, Toll-like receptor 4; ChIP, chromatin
immunoprecipitation; LPS, lipopolysaccharide. |
Discussion
In the present study, we investigated the gene
expression of LPS signaling receptor TLR4 in THP1 cells. The gene
expression of TLR4 was significantly increased in the presence of
high LPS concentration. The results were consistent with the
hypothesis that TLR4 is essential in LPS signaling. In
non-stimulated THP1 cells, TLR4 mRNA was constitutively expressed
and TLR4 may be essential to innate immunity in the first encounter
with gram-negative bacteria. The higher concentration and longer
constant LPS stimulation markedly promote TLR4 expression in the
THP1 cell membrane during the process of differentiation to
macrophages, indicating that a higher expression level of TLR4 has
the ability to lead a ‘cytokine storm’.
Our findings were based on the measurements of mRNA
and protein expression levels. However, the FACS results showed
that the surface expression of TLR4 in THP1 cell membranes was less
than the total TLR4 protein expression level. The surface
expression of TLR4 may not necessarily correlate with that of the
mRNA level, while the high LPS stimulation was able to supply
sufficient signaling to reinforce TLR4 accumulation in the cell
membrane.
TNF-α can upregulate TLR2 gene expression,
and the serum TNF-α level increases in response to LPS. Thus, the
same positive feedback mechanism may be present in the response of
TLR4 expression to LPS, and as a consequence, TLR4 would probably
contribute to accelerating immune responses in macrophages. If this
occurs, the regulation of TLR4 expression may be one of the immune
regulatory mechanisms commonly involved in host defense against
many bacterial strains. THP1 cells were selected to regulate TLR4
expression with a positive-feedback loop in a high concentration of
LPS for various reasons. First, according to the strength of
external signals, the expression intensity of the downstream
functional genes was induced by macrophages to prevent the invasion
of external antigens and not waste energy. Second, the accumulation
of TLR4 protein itself in THP1 cell membranes had the ability to
transmit increased LPS signals. Third, the form of
positive-feedback regulation of TLR4 signaling was efficient and
rapid. To demonstrate this positive positive-feedback mechanism,
the TLR4 inhibitor, CLI-095, was used to interrupt TLR4 signaling,
and in turn, TLR4 protein in the THP1 cell membrane was almost
depleted.
In macrophages, NF-κB activation seems to be
essential for LPS-mediated TLR4 induction. LPS is a strong
activator of NF-κB and all of the cytokines shown here that
increase TLR4 expression are known as activators of NF-κB (2,3).
Additionally, helenalin, an inhibitor of NF-κB transcriptional
activity at high concentrations, completely inhibited LPS-mediated
TLR4 induction at a concentration of 5 µmmol/l. Although helenalin
is not a specific inhibitor of NF-κB activation, the dose response
of TLR4 mRNA inhibition correlated well with that of NF-κB
activation. Furthermore, STA-21, a specific inhibitor of STAT3
transcriptional activity was only slightly inhibited to
LPS-mediated TLR4 induction. These results were noteworthy because
NF-κB and STAT3 were in sharp contrast with regard to binding to
TLR4 promoter in THP1 cells. This difference clearly suggested that
the regulatory roles of NF-κB pathways in TLR4 mRNA exhibit
conserved characteristics, while STAT3 may vary considerably in
different cell types.
In conclusion, the data show that the expression of
the LPS signaling receptor gene TLR4, was strictly regulated
by LPS concentration and stimulation during THP1 cell
differentiation into macrophages. TLR4 was constitutively expressed
and remained constant after various stimulations, including LPS.
However, a higher concentration of LPS significantly increased TLR4
expression. This suggests a positive-feedback mechanism. When
gram-negative bacteria invade the host, macrophages first recognize
LPS through the constitutively expressed TLR4, and then according
to the strength of LPS signaling, which is accumulated by TLR4
expression. Recent studies have indicated that TLR4 mRNA expression
in macrophages was decreased within a few hours of LPS treatment,
whereas we did not observe any decrease in TLR4 mRNA until 4 h
after the LPS treatment. This result may be due to the different
concentration of LPS and cell types.
Limitations also existed in the present study. This
study only revealed that NF-κB can inhibit TLR4 activity induced by
LPS from the cellular and molecular level. However, how to transfer
these findings to clinical applications remains unclear. In
addition, the results of the present study should be confirmed in
large-scale future studies.
Acknowledgements
The present study was supported by a grant from the
Outstanding Subject Leader Training Plan of Health Bureau of
Shanghai Pudong New Area (no. PWRd 2013-11).
References
|
1
|
Bellanti JA: Immunology. Proceedings of
the Annual Meeting of the Medical Section of the American Life
Insurance Association. American Life Insurance Association.
Washington, D.C.. pp. 16–30. 1975;
|
|
2
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lien E, Sellati TJ, Yoshimura A, Flo TH,
Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD,
et al: Toll-like receptor 2 functions as a pattern recognition
receptor for diverse bacterial products. J Biol Chem.
274:33419–33425. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zeromski J, Mozer-Lisewska I and Kaczmarek
M: Significance of toll-like receptors expression in tumor growth
and spreading: a short review. Cancer Microenviron. 1:37–42. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
West AP, Brodsky IE, Rahner C, Woo DK,
Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS and
Ghosh S: TLR signalling augments macrophage bactericidal activity
through mitochondrial ROS. Nature. 472:476–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Flo TH, Ryan L, Latz E, Takeuchi O, Monks
BG, Lien E, Halaas Ø, Akira S, Skjåk-Braek G, Golenbock DT, et al:
Involvement of toll-like receptor (TLR) 2 and TLR4 in cell
activation by mannuronic acid polymers. J Biol Chem.
277:35489–35495. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsuboi N, Yoshikai Y, Matsuo S, Kikuchi T,
Iwami K, Nagai Y, Takeuchi O, Akira S and Matsuguchi T: Roles of
toll-like receptors in C-C chemokine production by renal tubular
epithelial cells. J Immunol. 169:2026–2033. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pfarr KM, Fischer K and Hoerauf A:
Involvement of Toll-like receptor 4 in the embryogenesis of the
rodent filaria Litomosoides sigmodontis. Med Microbiol Immunol.
192:53–56. 2003.PubMed/NCBI
|
|
10
|
Lien E, Means TK, Heine H, Yoshimura A,
Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, et
al: Toll-like receptor 4 imparts ligand-specific recognition of
bacterial lipopolysaccharide. J Clin Invest. 105:497–504. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Barton GM and Medzhitov R: Toll-like
receptor signaling pathways. Science. 300:1524–1525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang G and Ghosh S: Toll-like
receptor-mediated NF-kappaB activation: a phylogenetically
conserved paradigm in innate immunity. J Clin Invest. 107:13–19.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anders HJ, Banas B and Schlöndorff D:
Signaling danger: toll-like receptors and their potential roles in
kidney disease. J Am Soc Nephrol. 15:854–867. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Territo MC and Cline MJ: Mononuclear
phagocyte proliferation, maturation and function. Clin Haematol.
4:685–703. 1975.PubMed/NCBI
|
|
15
|
Suzuki Y, Ruiz-Ortega M, Lorenzo O,
Ruperez M, Esteban V and Egido J: Inflammation and angiotensin II.
Int J Biochem Cell Biol. 35:881–900. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wolf G, Wenzel U, Burns KD, Harris RC,
Stahl RA and Thaiss F: Angiotensin II activates nuclear
transcription factor-kappaB through AT1 and AT2 receptors. Kidney
Int. 61:1986–1995. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Leendertse M, Willems RJ, Giebelen IA, van
den Pangaart PS, Wiersinga WJ, de Vos AF, Florquin S, Bonten MJ and
van der Poll T: TLR2-dependent MyD88 signaling contributes to early
host defense in murine Enterococcus faecium peritonitis. J Immunol.
180:4865–4874. 2008. View Article : Google Scholar : PubMed/NCBI
|