Introduction
Breast cancer is the most common type of cancer
diagnosed among women and, in America, one in eight women develop
breast cancer in their lifetime, with breast cancer accounting for
almost one in eight cases of cancer and being the second leading
cause of cancer-associated mortality among women (1). Although there have been advances in
breast cancer diagnosis and treatment, the molecular mechanisms
underlying breast cancer remain to be fully elucidated,
particularly regarding chemotherapy resistance (2).
It is known that cisplatin is one of the most potent
antitumor agents, exhibiting clinical activity against a wide
variety of solid types of tumor, including breast cancer (3). Cisplatin can interact with nuclear
DNA and mitochondrial DNA (mtDNA) to form DNA adducts, primarily
intrastrand crosslink adducts, which activate several signal
transduction pathways, including those involving ATR, p53, p73 and
MAPK, which culminate in the activation of apoptosis (4–7).
However, clinical studies have shown that patients with estrogen
receptor (ER)-positive cancer are more likely to develop cisplatin
resistance (8,9). These findings suggest that estrogen
may protect breast cancer cell from cisplatin insult through
activating ER.
Previous studies have shown that estrogen can bind
to ERα and β to stimulate the transcription of nuclear respiratory
factor 1 (NRF1), and the NRF1 transcription factor can regulate the
transcription of mitochondrial transcription factor A (TFAM)
(10,11). TFAM is essential in mtDNA
replication and repair (12,13).
It is known that cisplatin can destroy mtDNA to trigger cell death
signaling (14). Therefore, the
present study hypothesized that estrogen may activate ER and then
elevate the expression of TFAM to promote cell survival from
cisplatin treatment.
Materials and methods
Patients and ethics statement
A total of 16 fresh ER-positive breast cancer
tissues and 16 triple-negative breast cancer tissues were collected
from patients with pathologically and clinically confirmed breast
cancer. The tissues were collected from patients with
pathologically and clinically confirmed breast cancer. The age of
these women patients ranged from 27 to 55 years old, and were
collected from the November 2012 to December 2014, and the sizes
ranged from 0.2×0.7 cm to 2.4×2.6 cm. All human tumor tissues were
obtained with written informed consent from the patients. The
Institutional Review Board of Cangzhou Central Hospital (Cangzhou,
China) approved the use of the tumor samples in the present
study.
Cell culture
MCF-7, T47D, MDA-MB-231 and MDA-MB-453 cell lines
were purchased from America Type Culture Collection (ATCC;
Manassas, VA, USA). All cells were maintained under standard
culture conditions of 37°C and 5% CO2 in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.), as
recommended by ATCC.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was purified from homogenized breast
cancer tissues or cells using TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc., waltham, MA, USA) following the
manufacturer's protocol. The RNA (2 µg) was reverse transcribed
using SuperScript Reverse Transcriptase III (Invitrogen; Thermo
Fisher Scientific, Inc.). qPCR analysis was performed using SYBR
green Supermix (ABI; Thermo Fisher Scientific, Inc.) in an ABI 7900
PCR system (ABI; Thermo Fisher Scientific, Inc.). All the
procedures were performed according to the manufacturer's
recommended protocol. Reactions were incubated at 95°C for 30 sec,
followed by 40 cycles at 95°C for 5 sec and 60°C for 31 sec, and
finally 95°C for 15 sec, 60°C for 1 min and 95°C for 15 sec. The
relative expression of NRF1 and TFAM was calculated using the
2−ΔΔCq method (15)
with GAPDH RNA as the reference gene. The housekeeping gene, GAPDH,
was used as an internal standard. The primers using in the present
study are listed in Table I.
 | Table I.Sequence of primers and shRNAs. |
Table I.
Sequence of primers and shRNAs.
| Primer/shRNA | Sequence (5′-3′) |
|---|
| NRF1 | F:
GGTCGCAGTCTCCACGG |
|
| R:
ATGTTCGGTTTGGGTCACTC |
| GAPDH | F:
AGCCTCAAGATCATCAGCAATGCC |
|
| R:
TGTGGTCATGAGTCCTTCCACGAT |
| TFAM | F:
TCCTCTCCAAAATGCCAGAG |
|
| R:
TCCAGTTTTCCTTTACAGTCTTCAG |
| Sh-#1 |
CCGGCGTGAGTATATTGATCCAGAACTCGAGTTCTGGATCAATATACTCACGTTTTTG |
| Sh-#2 |
CCGGGTAAGTTCTTACCTTCGATTTCTCGAGAAATCGAAGGTAAGAACTTACTTTTTG |
Western blot analysis
The cells were lysed in WB/IP lysis buffer (cat. no.
P0013; Beyotime Institute of Biotechnology, Haimen, China) and
nuclear proteins were extracted using lysis buffer (cat. no. P0028;
Beyotime Institute of Biotechnology). All procedures were performed
following the manufacturer's protocol. Protein concentrations were
determined by bicinchoninic acid assay. Subsequently, the cell
lysates were boiled in 5X SDS-PAGE loading buffer for 10 min, 30–50
µg samples were resolved by 8% SDS-PAGE and then transferred onto
nitrocellulose membranes. The membranes were incubated with the
following antibodies at 4°C overnight (1:1,000 dilution): NRF1
(cat. no. 12482-1-AP; ProteinTech Group, Inc.; Chicago, IL, USA),
TFAM (cat. no. 19998-1-AP; ProteinTech Group, Inc.), ERα (cat. no.
21244-1-AP; ProteinTech Group, Inc.) and GAPDH (cat. no. 2118; CST
Biological Reagents Company Limited, Shanghai, China).
Subsequently, the membranes were incubated with anti-rabbit (cat.
no. 5127) and anti-mouse (cat. no. 58802) secondary antibodies
(1:2,000 dilution; Cell Signaling Technology, Inc., Danvers, MA,
USA). The bound antibodies were visualized with an ECL kit (Thermo
Fisher Scientific, Inc.).
Construction of stable cell lines
To generate stable TFAM-silenced cell lines, vectors
containing short hairpin (sh)RNAs were purchased from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). 293T cells
(ATCC) were transfected with these vectors using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) with all procedures
performed according to the manufacturer's protocols. The
supernatant media containing the virus was collected by
centrifugation (1.2×105 × g for 10 min) to remove
cellular debris. The viruses were used to infect the indicated
cells, and the transfected cells were then selected by exposure to
2 µg/ml puromycin for 2 weeks. The alterations of TFAM in these
cells were confirmed using PCR and western blot analysis prior to
further analysis. The sequences of the shRNAs used are listed in
Table I. As Sh-# 2 showed a lower
relative efficiency, Sh-#1 was selected to perform the subsequent
experiments.
Overexpression of TFAM
The constructed stable cell lines were transfected
with pCDNA3.1 (Thermo Fisher Scientific, Inc.), which contained the
open reading frame of TFAM, using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) to re-introduce TFAM to these cell
lines. All procedures were performed according to the
manufacturer's protocol.
Cell Counting Kit-8 (CCK-8) cell
viability assays and caspase 3/7 apoptosis assays
The cells were seeded into a 96-well plate at a
density of 3×103 cells per well with 100 µl culture
medium and MCF-7 cells were seeded in 6-well plates at a density of
300,00 cells/well, and then cells were placed in phenol-red media
supplemented with 5% dextran-charcoal-stripped fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), MEM nonessential amino
acids (Gibco; Thermo Fisher Scientific, Inc.), gentamicin (MCE
China, Shanghai, China) and 6 ng/ml bovine insulin (YuanYe
Biotechnology Co., Ltd., Shanghai, China) for 48 h at 37°C. The
cells were then treated with estradiol (0.1 nM; Selleck Chemicals,
Houston, TX, USA) or estradiol and fulvestrant (6 µg/ml; Selleck
Chemicals) for 24 h at 37°C, following which and the medium was
replaced and the cells were treated with the indicated
concentrations (0, 2, 5, 10, 15 and 20 µM) of cisplatin
(Sigma-Alrich; Merck Millipore) for 14 h at 37°C. The cell
viabilities were then quantified by the addition 10 µl of CCK-8
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). Following
incubation for 1.5 h at 37°C, the plates were monitored using a
Power Wave XS microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA) at an absorbance 450 nm. Cell apoptosis assays
were performed using a Caspase3/7 Glo kit (Promega Corporation,
Madison, WI, USA) and all procedures were performed according to
the manufacturer's protocols.
Tumorigenesis in vivo
All mice (n=48) were purchased from Shanghai
Laboratory Animal Center (Shanghai, China). All the mice were 8
week-old females and were housed at 25°C with access to sterile
water and food. A total of 1.0×106 of the stably
transfected MCF-7 cells (Sh-#1 or negative control shRNA) were
implanted subcutaneously into the right flank of BALB/c (nu/nu)
mice. At the same time, estradiol (0.06 mg) or estradiol and
fulvestrant (0.06 mg) were injected every 2 days for 2 months
(eight in each treatment group). After the 2 months, cisplatin was
injected at a dose of 10 mg/kg body weight every 2 days for 2
months. After 8 weeks, the mice were sacrificed by cervical
dislocation for the analysis of tumor burden. All procedures were
performed in accordance with The Animal Care and Use Committee of
the Second Military Medical University (Shanghai, China).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Analysis was performed using SPPS software (version 19; IBM SPSS,
Armonk, NY, USA) Two tail student's t-test was used for comparisons
between control and test groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
ER-positive breast cancer cells are
more sensitive to cisplatin
Clinical studies have shown that patients with
triple-negative breast cancer are more sensitive to cisplatin
chemotherapy, compared with patients with non-triple-negative
breast cancer, particularly those with ER-positive cancer (16). This suggests that ER may be
essential in cisplatin chemoresistance, and the present study was
performed to investigate the underlying mechanism. In the present
study, ER-positive MCF-7 and T47D breast cancer cell lines, and
ER-negative MDA-MB-231 and MDA-MB-453 breast cancer cell lines
(17–20) were selected to detect their
sensitivies to cisplatin. All cell lines were treated with the
indicated concentrations of cisplatin for 24 h, and their
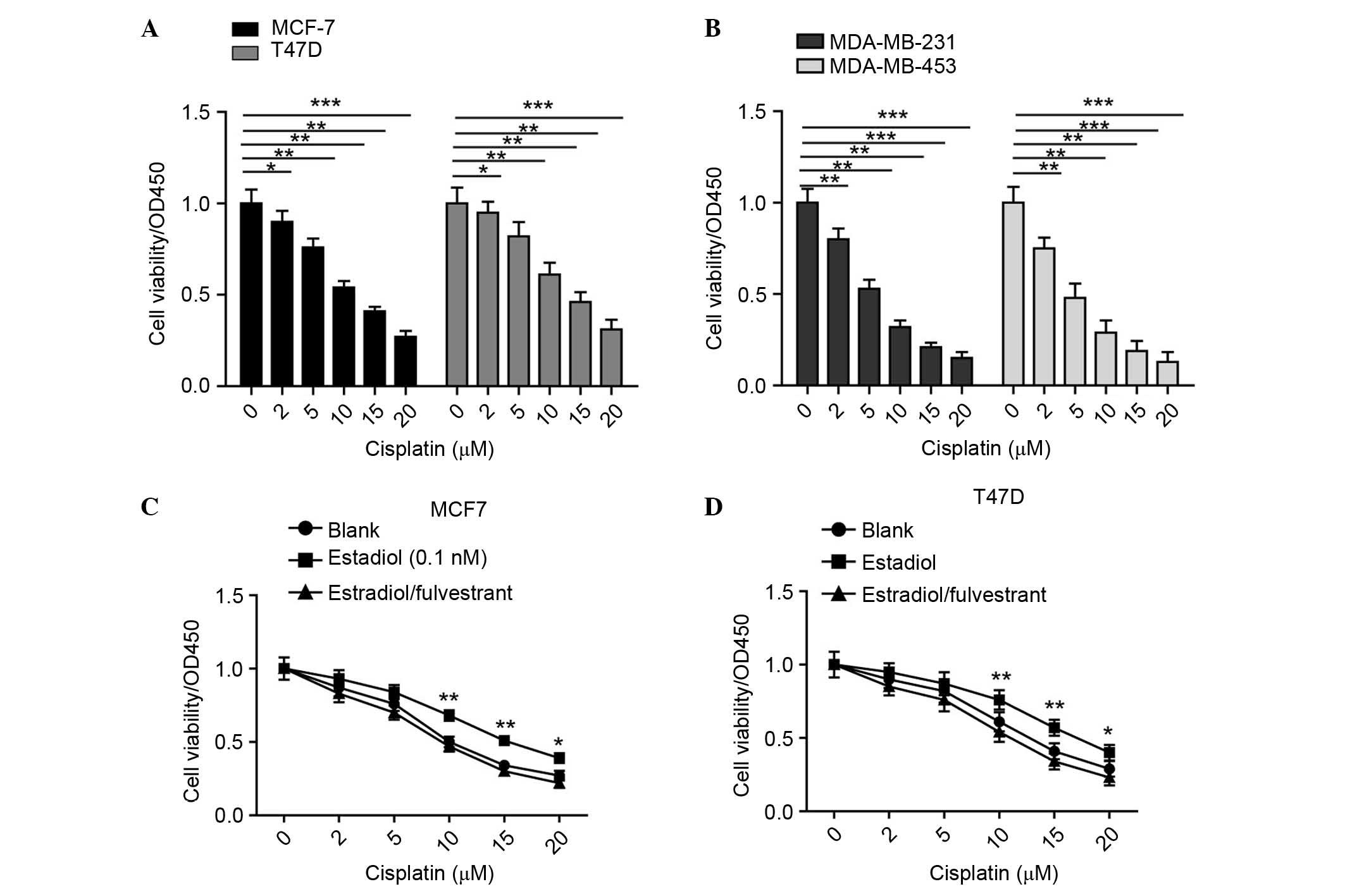
viabilities were examined using CCK-8 assays. As shown in Fig. 1A and B, the results confirmed the
clinical observation; ER-negative cells were more sensitive to
cisplatin treatment. These findings suggested that ER has an
aggressive role in the chemoresistance of cells to cisplatin.
Subsequently, MCF-7 and MDA-MB-231 cell lines were selected to
examine the reactions when the cells were treated with estradiol or
with estradiol and fulvestrant, an ER inhibitor. As shown in
Fig. 1C and D, it was found that,
when treated with estradiol, the MCF-7 and T47D cells exhibited a
higher tolerance to the different concentrations of cisplatin, and
this increased tolerance was reversed when the cells were incubated
with estradiol and fulvestrant. Taken together, these results
confirmed the oncogenic role of ER in cisplatin resistance in
breast cancer.
Levels of TFAM are positively
correlated with NRF1 and ER
The mechanism underlying the effect of ER in
cisplatin resistance in breast cancer remains to be fully
elucidated. A previous report provided evidence to suggest that
NRF1 may be essential during the chemoresistance process in breast
cancer (21). In the present
study, the mRNA level of NRF1 was detected in 18 ER-positive
patient tumor tissues and 18 ER-negative tissues. As shown in
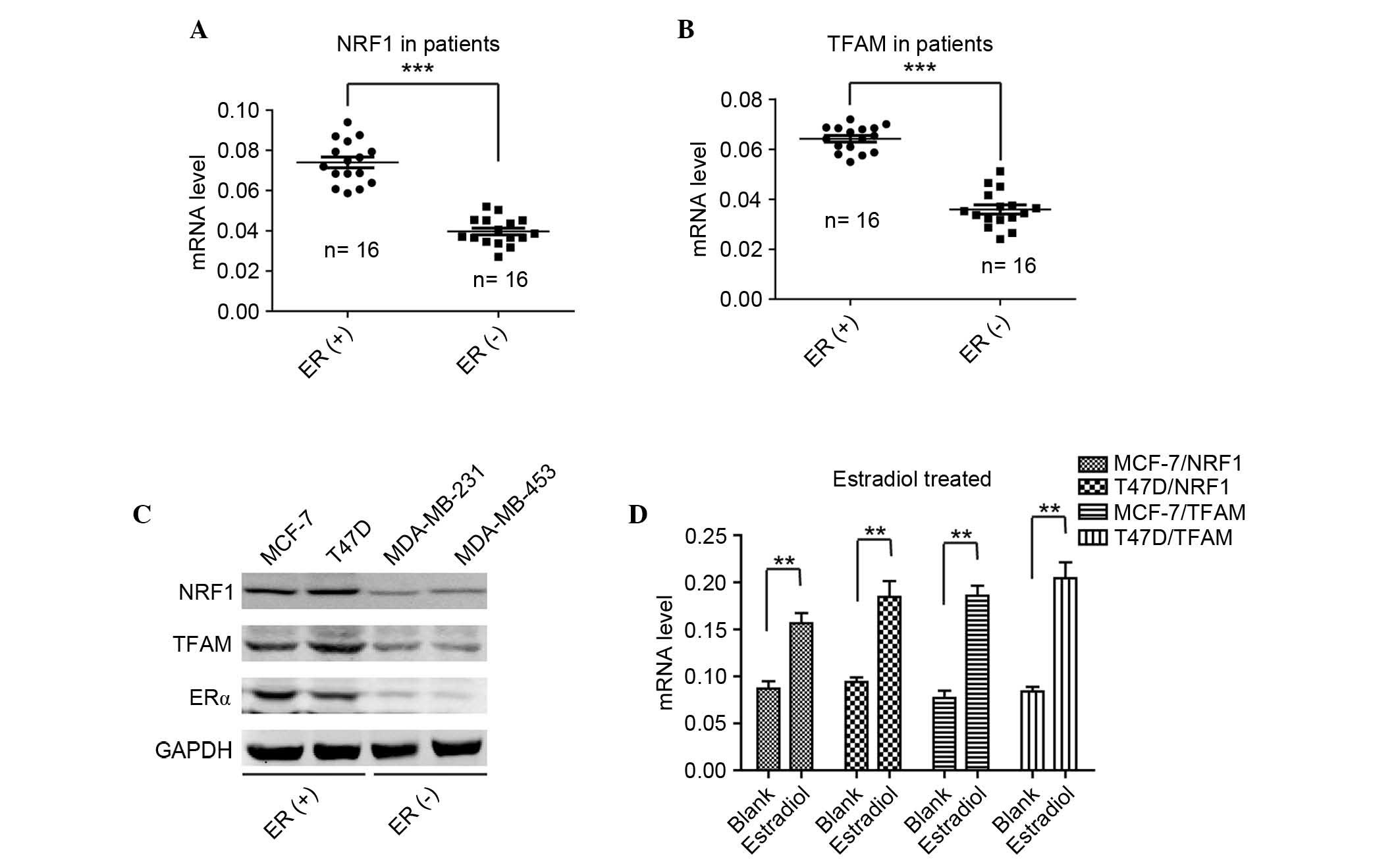
Fig. 2A, the expression levels of
NRF1 were significantly higher in the ER-positive tissues, compared
with the ER-negative tissues. The expression pattern of NRF1 in the
breast cancer tissues prompted investigation to further elucidate
the role of NRF1 in cisplatin resistance in breast cancer. NRF1
functions as a transcription factor and previous reports on the
functions of NRF1 have revealed that TFAM, which encodes a key
mitochondrial transcription factor, and is essential in mtDNA
replication and repair, is usually transactivated by NRF1 (11). As is already known, cisplatin
functions to destroy the double strand of DNA and mtDNA is usually
damaged by cisplatin (22). The
present study hypothesized that a high expression level of TFAM may
protect ER-positive breast cancer cells from the insult of
cisplatin (4). The present study
detected the mRNA levels of TFAM in the same two groups of breast
cancer tissues, and found that the levels of TFAM in the
ER-positive tissues were significantly higher, compared with those
in the ER-negative tissues (Fig.
2B). Furthermore, it was found that the expression of TFAM was
positively correlated with the level of NRF1 in the ER-positive
breast cancer tissues (Fig. 2B).
To confirm the results from the breast cancer tissues, the present
study examined the protein levels of NRF1, TFAM and ERα in the
ER-positive and ER-negative cell lines. As shown in Fig. 2C, it was found that the protein
levels of NRF1 and TFAM were positively correlated with the
expression of ERα, and that NRF1 and TFAM were expressed at high
levels in the MCF-7 and T47D cells, and relatively lower in the
MDA-MB-231 and MDA-MB-453 cells. To further validate the
associations among ER, NRF and TFAM, the ER-positive MCF-7 and T47D
cells were selected to examine the alterations when treated with
estradiol. As shown in Fig. 2D,
the levels of NRF1 and TFAM mRNA were significantly elevated when
the cells were treated with estradiol. These findings indicated
that estradiol activated ER and then transactivated the
transcription of NRF1, followed by the elevated expression of TFAM
to promote ER-positive breast cancer cell survival from cisplatin
therapy.
TFAM knockdown elevates sensitivity to
cisplatin in ER-positive breast cancer cells
The present study further investigated whether TFAM
regulates the sensitivity of ER-positive breast cancer cell to
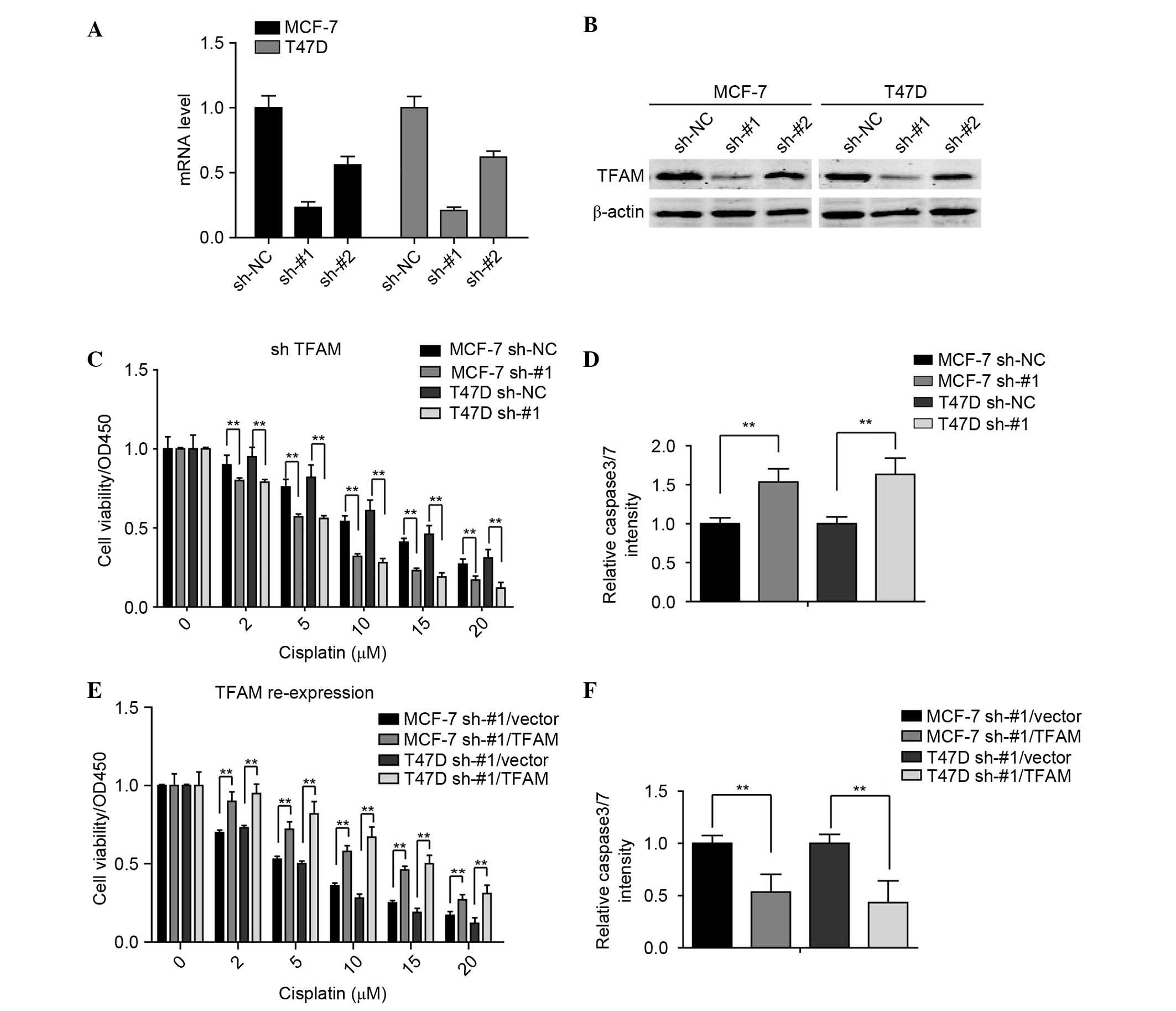
cisplatin. For this investigation, two pairs of shRNAs (sh-#1 and
sh-#2) specific against TFAM, were used to generate stable TFAM
knockdown and control shRNA transfection in the MCF-7 and T47D cell
lines. The results showed that sh-#1 effectively knocked down TFAM
in these breast cancer cell lines (Fig. 3A and B).
The present study then examined the effect of TFAM
knockdown on breast cancer cell viability when treated with
different concentrations of cisplatin. As shown in Fig. 3C, TFAM knockdown significantly
inhibited the cell viabilities. The activities of caspase 3/7 in
these stable cell lines were also detected, and it was found that
the cells with TFAM knockdown exhibited higher relative activities
of caspase 3/7 (Fig. 3D).
Furthermore, to demonstrate that the effects on the were
specifically due to the silencing of TFAM, TFAM was reintroduced
into the sh-#1-transfected MCF-7 and T47D cells, and it was found
that the re-expression of TFAM reversed the TFAM knockdown-induced
cisplatin insult (Fig. 3E and F).
Taken together, these findings indicated that TFAM may be closely
associated with the sensitivity of breast cancer cells to
cisplatin.
TFAM knockdown elevates sensitivity to
cisplatin in ER-positive breast cancer cells in vivo
The effects of TFAM knockdown in the breast cancer
cells prompted the investigation of whether TFAM had the same
effect in vivo. The present study used stably-transfected
MCF-7 cell lines and performed xenograft tumor growth assays. As
shown in Fig. 4A and B, it was
found that the knockdown of TFAM resulted in the tumor size and
weight being significantly smaller and lighter, respectively,
compared with the control groups when treated with estradiol. These
in vivo findings confirmed that TFAM was essential in
cisplatin resistance in the ER-positive breast cancer.
Discussion
In the present study, it was found that TFAM
promoted ER-positive breast cancer cell survival following
cisplatin treatment. Previously, it has been shown that patients
with ER-positive breast cancer are more likely to exhibit the
features of cisplatin resistance (23). The present study provided
preliminary clues to the molecular mechanism underlying this
clinical observation.
Regarding the sensitivity of breast cancer cells to
cisplatin, the findings of the present study were consistent with
previous evidence showing that ER-negative breast cancer cells are
more sensitive to cisplatin treatment, and ER-positive breast
cancer cells are more tolerant to cisplatin (24). It is known that several oncogenic
genes and tumor suppressors are associated with cisplatin
sensitivity, including AKT, c-myc and p53 (25–27);
however, cisplatin resistance is a cell type- and tissue
dependent-phenomenon, and the clinical mechanisms leading to
cisplatin resistance are complex (28–30).
In the present study, it was found that estrogen indirectly
regulated TFAM, which may also be involved in cisplatin
resistance.
ER can be activated by estrogen, and activated ER
can activate the transcription of NRF1, which can regulate the
expression of TFAM. TFAM is a mitochondrial transcription factor,
which functions in mitochondrial transcription regulation, and also
functions in mitochondrial DNA replication and repair (10,11).
As the molecular mechanism underlying the effect of cisplatin on
cancer cell death is to disturb the double strand of DNA, including
mtDNA (31), these previous
studies indicate a potential mechanism by which TFAM can protect
mtDNA from cisplatin-induced injury.
The in vitro and in vivo experiments
performed in the present study confirmed the hypothesis that TFAM
has a protective effect in ER-positive breast cancer cells exposed
to cisplatin treatment. It is known that mitochondrial damage is a
major trigger for the induction of cell apoptosis and necrosis
(32), however, the detailed role
of TFAM remains to be fully elucidated and requires further
investigation.
In conclusion, the present study demonstrated that
estrogen indirectly regulated the expression of TFAM to protect
ER-positive breast cancer cells from cisplatin treatment. These
results offer potential in guiding chemotherapy in patients with
breast cancer.
Acknowledgements
This study was supported by the Shanghai Science and
Technology Committee (grant no. 14ZR1408800).
References
|
1
|
DeSantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kwok JM, Peck B, Monteiro LJ, Schwenen HD,
Millour J, Coombes RC, Myatt SS and Lam EW: FOXM1 confers acquired
cisplatin resistance in breast cancer cells. Mol Cancer Res.
8:24–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang J, Wang Z, Hu X, Wang B, Wang L,
Yang W, Liu Y, Liu G, Di G, Hu Z, et al: Cisplatin and gemcitabine
as the first line therapy in metastatic triple negative breast
cancer. Int J Cancer. 136:204–211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yazlovitskaya EM and Persons DL:
Inhibition of cisplatin-induced ATR activity and enhanced
sensitivity to cisplatin. Anticancer Res. 23:2275–2279.
2003.PubMed/NCBI
|
|
6
|
Wei Q, Dong G, Yang T, Megyesi J, Price PM
and Dong Z: Activation and involvement of p53 in cisplatin-induced
nephrotoxicity. Am J Physiol Renal Physiol. 293:F1282–F1291. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshida K, Ozaki T, Furuya K, Nakanishi M,
Kikuchi H, Yamamoto H, Ono S, Koda T, Omura K and Nakagawara A:
ATM-dependent nuclear accumulation of IKK-alpha plays an important
role in the regulation of p73-mediated apoptosis in response to
cisplatin. Oncogene. 27:1183–1188. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pegram MD, Lipton A, Hayes DF, Weber BL,
Baselga JM, Tripathy D, Baly D, Baughman SA, Twaddell T, Glaspy JA
and Slamon DJ: Phase II study of receptor-enhanced chemosensitivity
using recombinant humanized anti-p185HER2/neu monoclonal antibody
plus cisplatin in patients with HER2/neu-overexpressing metastatic
breast cancer refractory to chemotherapy treatment. J Clin Oncol.
16:2659–2671. 1998.PubMed/NCBI
|
|
9
|
Sui M, Zhang H and Fan W: The role of
estrogen and estrogen receptors in chemoresistance. Curr Med Chem.
18:4674–4683. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mattingly KA, Ivanova MM, Riggs KA,
Wickramasinghe NS, Barch MJ and Klinge CM: Estradiol stimulates
transcription of nuclear respiratory factor-1 and increases
mitochondrial biogenesis. Mol Endocrinol. 22:609–622. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cam H, Balciunaite E, Blais A, Spektor A,
Scarpulla RC, Young R, Kluger Y and Dynlacht BD: A common set of
gene regulatory networks links metabolism and growth inhibition.
Mol Cell. 16:399–411. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang D, Kim SH and Hamasaki N:
Mitochondrial transcription factor A (TFAM): Roles in maintenance
of mtDNA and cellular functions. Mitochondrion. 7:39–44. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hallberg BM and Larsson NG: TFAM forces
mtDNA to make a U-turn. Nat Struct Mol Biol. 18:1179–1181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Z, Schumaker LM, Egorin MJ, Zuhowski
EG, Guo Z and Cullen KJ: Cisplatin preferentially binds
mitochondrial DNA and voltage-dependent anion channel protein in
the mitochondrial membrane of head and neck squamous cell
carcinoma: Possible role in apoptosis. Clin Cancer Res.
12:5817–5825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Byrski T, Huzarski T, Dent R, Gronwald J,
Zuziak D, Cybulski C, Kladny J, Gorski B, Lubinski J and Narod SA:
Response to neoadjuvant therapy with cisplatin in BRCA1-positive
breast cancer patients. Breast Cancer Res Treat. 115:359–363. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hsieh CY, Santell RC, Haslam SZ and
Helferich WG: Estrogenic effects of genistein on the growth of
estrogen receptor-positive human breast cancer (MCF-7) cells in
vitro and in vivo. Cancer Res. 58:3833–3838. 1998.PubMed/NCBI
|
|
18
|
Ström A, Hartman J, Foster JS, Kietz S,
Wimalasena J and Gustafsson JA: Estrogen receptor beta inhibits
17beta-estradiolstimulated proliferation of the breast cancer cell
line T47D. Proc Natl Acad Sci USA. 101:1566–1571. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferguson AT, Lapidus RG, Baylin SB and
Davidson NE: Demethylation of the estrogen receptor gene in
estrogen receptor-negative breast cancer cells can reactivate
estrogen receptor gene expression. Cancer Res. 55:2279–2283.
1995.PubMed/NCBI
|
|
20
|
Doane AS, Danso M, Lal P, Donaton M, Zhang
L, Hudis C and Gerald WL: An estrogen receptor-negative breast
cancer subset characterized by a hormonally regulated
transcriptional program and response to androgen. Oncogene.
25:3994–4008. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li L and Yu AQ: The functional role of
peroxiredoxin 3 in reactive oxygen species, apoptosis, and
chemoresistance of cancer cells. J Cancer Res Clin Oncol.
141:2071–2077. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gonzalez-Angulo AM, Morales-Vasquez F and
Hortobagyi GN: Overview of resistance to systemic therapy in
patients with breast cancer. Adv Exp Med Biol. 608:1–22. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Silver DP, Richardson AL, Eklund AC, Wang
ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, et
al: Efficacy of neoadjuvant Cisplatin in triple-negative breast
cancer. J Clin Oncol. 28:1145–1153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Funato T, Kozawa K, Kaku M and Sasaki T:
Modification of the sensitivity to cisplatin with c-myc
over-expression or down-regulation in colon cancer cells.
Anticancer Drugs. 12:829–834. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fraser M, Bai T and Tsang BK: Akt promotes
cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function. Int J
Cancer. 122:534–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bradford CR, Zhu S, Ogawa H, Ogawa T,
Ubell M, Narayan A, Johnson G, Wolf GT, Fisher SG and Carey TE: P53
mutation correlates with cisplatin sensitivity in head and neck
squamous cell carcinoma lines. Head Neck. 25:654–661. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niedner H, Christen R, Lin X, Kondo A and
Howell SB: Identification of genes that mediate sensitivity to
cisplatin. Mol Pharmacol. 60:1153–1160. 2001.PubMed/NCBI
|
|
29
|
Funato T, Kozawa K, Kaku M and Sasaki T:
Modification of the sensitivity to cisplatin with c-myc
over-expression or down-regulation in colon cancer cells.
Anticancer Drugs. 12:829–834. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fraser M, Bai T and Tsang BK: Akt promotes
cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function. Int J
Cancer. 122:534–546. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olivero OA, Semino C, Kassim A,
Lopez-Larraza DM and Poirier MC: Preferential binding of cisplatin
to mitochondrial DNA of Chinese hamster ovary cells. Mutat Res.
346:221–230. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kroemer G, Dallaporta B and Resche-Rigon
M: The mitochondrial death/life regulator in apoptosis and
necrosis. Annu Rev Physiol. 60:619–642. 1998. View Article : Google Scholar : PubMed/NCBI
|