Introduction
Cigarette smoke contains >4,000 toxic chemicals
with hazardous adverse effects on almost all organs in the body
(1). The major clinical
consequences of cigarette smoking include chronic respiratory
ailments, increased incidence of several types of cancer and
increased risk of cardiovascular disorders (2,3). In
addition, smoking may accelerate the progression of fibrosis in
patients with chronic renal or pancreatic diseases (4,5).
Although not considered a causative agent, smoking may have a
negative effect on the incidence, severity and clinical course of
liver diseases, including primary biliary cirrhosis, chronic
hepatitis C infection, nonalcoholic fatty liver disease and
hepatocellular carcinoma (6–9). The
mechanisms underlying smoking-induced liver alterations are
complicated and remain to be fully elucidated.
The liver is the critical site in the body for the
removal of toxins, and process alcohol and drugs. To perform these
functions, the liver expresses heme-containing enzymes of the
cytochrome P450 (CYP) families (10). In humans, the CYP1, CYP2 and CYP3
families are involved in hepatic drug metabolism. Other CYP
families are involved in the biosynthesis and metabolism of
steroids and retinoic acid. The CYP1 family has three members,
CYP1A1, CYP1A2 and CYP1A3. CYP1A2 is expressed predominantly in the
liver and is responsible for the metabolism of several drugs,
including theophylline, which is used clinically to prevent and
treat the wheezing, shortness of breath and chest tightness caused
by asthma, chronic bronchitis, emphysema and other lung diseases
(11). In the CYP2 family, the
CYP2C subfamily constitutes ~20% of the total hepatic CYP and is
involved in the metabolism of a wide variety of drugs in clinical
use (12). Although CYP2D6
represents only 2% of the hepatic CYP content in humans, it
metabolizes >70 drugs (11).
Among the CYP3 family members, CYP3A4 is primarily expressed in the
liver and catalyzes the metabolism of a number of drugs, for
example statins, which are used to treat hyperlipidemia (13). The expression of CYP is considered
to be controlled by nuclear factor (NF)-κB through nuclear
receptors (14). Pregnane X
receptor (PXR) can recognize and bind to the responsive elements in
CYP2B and CYP2C genes (15).
Constitutive androstane receptor (CAR), a nuclear receptor, has
been shown to be involved in the regulation of CYP2C9 and CYP3A4
(16,17). Glucocorticoid responsive element
has been identified in the regulatory region of the CYP2C9 promoter
(18). The activation of
glucocorticoid receptor (GR) is essential for the induction of
CYP3A4 by glucocorticoids (19).
The activation of NF-κB is affected by
pro-inflammatory cytokines, including interleukin 6 (IL6),
interleukin 1 (IL1), and tumour necrosis factor α (TNFα), which are
promoted by chemicals in cigarette smoke (20). The development of inflammation is
regulated by autophagy through a chain of elements, including
autophagy-related protein 5 (ATG5) and ATG12 (21,22).
The in vitro treatment of liver cells with ethanol has been
shown to increase the expression of IL6, an effect which was
markedly alleviated by rapamycin, an inducer of autophagy (23). However, whether autophagic activity
is altered in the liver of cigarette smokers remains to be
elucidated.
In the present study, a rat model of smoking was
established to examine the effects of cigarette smoking on
inflammation, autophagic activity, and the expression of nuclear
receptor and CYP in the liver. It was found that smoking induced a
reduction in the expression of drug-metabolizing CYPs in the liver
through the regulation of nuclear receptors. These findings
indicate the importance of considering metabolic ability in the
liver of patients who smoke prior to prescribing drugs.
Materials and methods
Ethics statement
The present study was performed in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals of Tianjin Medical University (Tianjin, China).
The protocol was approved by the Animal Care Committee of Tianjin
Medical University (Permit no. 2010-0002). All surgery was
performed under sodium pentobarbital anesthesia, and all efforts
were made to minimize suffering of animals.
Reagents
TRIzol reagent was purchased from Invitrogen; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). The TIANScript RT kit
was purchased from Tiangen Biotech Co., Ltd. (Beijing, China). SYBR
Green polymerase chain reaction (PCR) core reagents were purchased
from Bio-Rad Laboratories, Inc. (Hercules, CA, USA). RPMI 1640
medium was purchased from Gibco; Thermo Fisher Scientific, Inc.
Fetal calf serum was purchased from Hyclone; GE Healthcare Life
Sciences (Logan, UT, USA). Dexamethasone (Dex) was purchased from
Sigma-Aldrich; Merck Millipore (Billerica, MA, USA). RU486
(mifepristone), a specific antagonist of glucocorticoid receptor,
was purchased from Sigma-Aldrich; Merck Millipore.
Animals and treatment
Male Wistar rats weighing 180±20 g and aged 6 weeks
were purchased from the Model Animal Center of the Radiological
Medicine Research Institute, Chinese Academy of Medical Science
(Beijing, China). The rats were housed in standard laboratory cages
(n=5) at 22°C with a 12 h light/dark cycle and free access to food
and water. A total of 30 rats were divided into two groups (15 per
group), comprising a cigarette smoking-exposed group and an
unexposed control group. As described previously (24), the smoking group received
whole-body exposure to the smoke of five unfiltered cigarettes
(Daqianmen™; tar ≤15 mg, nicotine ≤1.1 mg and CO ≤13 mg) for 30
min, twice daily (prior to 9:00 a.m and after 5:00 p.m), for 14
weeks inside a 0.6 m3 custom plexiglass chamber
(constructed in-house). The protocol for the control group was
identical, but without smoke exposure.
Cell culture
LO2 cells (a gift from Chenghu Liu lab, NanKai
University, Tianjin, China) were maintained in RPMI 1640 medium
supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C with 5%
CO2. In the experiments, 10 nM Dex and 10 µM RU486 were
added into the culture medium when the cells were 80%
confluence.
Liver tissue sampling
At the end of smoking exposure period, the rats were
anesthetized with sodium pentobarbital and sacrificed by cervical
dislocation. The abdominal cavity was opened and the liver tissues
were excised, rinsed in ice-cold PBS (pH 7.4), and then either
stored at −80°C for the analysis of gene expression or fixed in 10%
neutral-buffered formalin for the analysis of histology.
Hematoxylin and eosin (HE)
staining
Following fixation in 10% neutral-buffered formalin,
the liver tissues were embedded in paraffin and 5 µm thick sections
were cut. The sections were then stained with HE solution (Solarbio
Science & Technology Co., Ltd., Beijing, China) and images of
the staining were captured under an Olympus IX71 microscope with a
DP80 camera (Olympus Corporation, Tokyo, Japan).
Total RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR) analysis
RNA was extracted from either the liver tissues or
the LO2 cells using TRIzol reagent. The RNA (3 µg) was then reverse
transcribed using oligo (dT) primers for 1 h at 50°C using the
TIANScript RT kit (Tiangen Biotech, Co., Ltd.) according to the
manufacturer's protocol. The qPCR analysis was performed using the
SYBR Green method. Specific gene primers were designed using
Primer-Quest SM software (http://www.idtdna.com/Scitools/Applications/PrimerQuest;
Integrated DNA Technologies, Inc., Coralville, IA, USA), and then
commercially produced (BGI Tech, Shenzhen, China; listed in
Tables I and II). The DNA amplification reactions were
performed on a Light Cycler 96 real-time PCR system (Roche
Diagnostics, Indianapolis, IN, USA) under the following reaction
conditions: An initial heating cycle of 95°C for 2 min; and 40
cycles alternating between denaturation at 95°C for 25 sec, primer
annealing at 60°C for 25 sec and extension at 72°C for 20 sec.
Melting curves were used to clarify the identity of amplicons, and
the housekeeping gene, GADPH, served as an internal control. The
relative mRNA expression levels of targeted genes were calculated
using the comparative Cq (quantification cycle) method normalized
to GAPDH mRNA in the same sample. Briefly, specific ΔCq was
calculated as follows: ΔCq = (CqGAPDH) - (Cqtarget); and relative
expression was defined as: 2−ΔCq (24).
 | Table I.Sequences of primers for genes
expressed in rats. |
Table I.
Sequences of primers for genes
expressed in rats.
| Gene | Direction | Sequence
(5′-3′) |
|---|
| IL-1β | Forward |
TCCCTGAACTCAACTGTGAAATA |
|
| Reverse |
GGCTTGGAAGCAATCCTTAATC |
| IL-6 | Forward |
GAAGTTAGAGTCACAGAAGGAGTG |
|
| Reverse |
GTTTGCCGAGTAGACCTCATAG |
| TNF-α | Forward |
ACCTTATCTACTCCCAGGTTCT |
|
| Reverse |
GGCTGACTTTCTCCTGGTATG |
| PXR | Forward |
GAAGATCATGGCTGTCCTCAC |
|
| Reverse |
CGTCCGTGCTGCTGAATAA |
| CAR | Forward |
GAGACCATGACCAGTGAAGAAG |
|
| Reverse |
AGTCAGGGCATGGAAATGATAG |
| GR | Forward |
CAGCAGTGAAATGGGCAAAG |
|
| Reverse |
GGGCAAATGCCATGAGAAAC |
| Ampk | Forward |
GCTGACTTCGGACTCTCTAATATG |
|
| Reverse |
CATACAGCCTTCCTGAGATGAC |
| Wip1 | Forward |
GTCTGGAGTGAATCGTGTAGTT |
|
| Reverse |
CTACGGCCAAGAAAGGAATCT |
| Atg5 | Forward |
TCCAACGTGCTTTACTCTCTATC |
|
| Reverse |
TGTCAGTTACCAGCGTCAAATA |
| Atg12 | Forward |
TGAAGGCTGTAGGAGACACT |
|
| Reverse |
GCCAGCAGTCTGAGGAATTT |
| Ulk1 | Forward |
GCAGTTGCTTCTGGCTCTAT |
|
| Reverse |
GGGTGCTGGCATCTAAGAAA |
| Anx3 | Forward |
CAGATGAAGACACCCTGATTGA |
|
| Reverse |
TCCAGACGTTTCAGAGCTAATG |
| Cyp1A2 | Forward |
GACAAGACCCTGAGTGAGAAG |
|
| Reverse |
GAGGATGGCTAAGAAGAGGAAG |
| Cyp2C9 | Forward |
CCCAAGGGCACAACCATATTA |
|
| Reverse |
CTTTCTGGATGAAGGTGGCA |
| Cyp2C19 | Forward |
CCCAAGGGCACAACCATATTA |
|
| Reverse |
TTTGACCCTCGTCACTTTCTG |
| Cyp2D4 | Forward |
CCTTTCAGCCCTAACACTCTAC |
|
| Reverse |
ATGAAGCGTGGGTCATTGT |
| Cyp3A2 | Forward |
GGAAACCCGTCTGGATTCTAAG |
|
| Reverse |
GAAGTGTCTCATAAAGCCCTGT |
| Gapdh | Forward |
ACTCCCATTCTTCCACCTTTG |
|
| Reverse |
AATATGGCTACAGCAACAGGG |
| Table II.Sequences of primers for genes
expressed in LO2 cells. |
Table II.
Sequences of primers for genes
expressed in LO2 cells.
| Gene | Direction | Sequence
(5′-3′) |
|---|
| hmTOR | Forward |
GGGACTACAGGGAGAAGAAGAA |
|
| Reverse |
GCATCAGAGTCAAGTGGTCATAG |
| hULK1 | Forward |
GTGGGCAAGTTCGAGTTCT |
|
| Reverse |
GACTTGGCGAGGTTCTTCTT |
| hATG5 | Forward |
GGAATTGAGCCAATGTTGGAAA |
|
| Reverse |
GTTGGCTGTGGGATGATACTAA |
| hATG12 | Forward |
CGTCTTCCGCTGCAGTTT |
|
| Reverse |
GGAAGGAGCAAAGGACTGATT |
| hATG13 | Forward |
CAAGCTCTCGCCTTTCCTATC |
|
| Reverse |
GGTGAGGGTGTGTAGCATTTA |
| hLC3 | Forward |
ACAGCATGGTGAGTGTGTC |
|
| Reverse |
GGGAGGCGTAGACCATATAGA |
| hBeclin 1 | Forward |
CCCGTGGAATGGAATGAGATTA |
|
| Reverse |
CCGTAAGGAACAAGTCGGTATC |
| hCYP1A2 | Forward |
CAGGAGCACTATCAGGACTTTG |
|
| Reverse |
CAATCTTCTCCTGTGGGATGAG |
| hCYP2C9 | Forward |
CCCAAGGGCACAACCATATTA |
|
| Reverse |
TGCCACCTTCATCCAGAAAG |
| hCYP2C19 | Forward |
CCCAAGGGCACAACCATATTA |
|
| Reverse |
CAGAAAGTGACGAGGGTCAAA |
| hCYP2D6 | Forward |
CATGGAGCTCTTCCTCTTCTTC |
|
| Reverse |
ACAGCACAAAGCTCATAGGG |
| hCYP3A4 | Forward |
GCTGAGGATGAAGAATGGAAGA |
|
| Reverse |
CTCCATACTGGGCAATGATAGG |
| hGADPH | Forward |
GGTGTGAACCATGAGAAGTATGA |
|
| Reverse |
GAGTCCTTCCACGATACCAAAG |
Statistical analysis
The data were analyzed using SPSS software, version
13.0 (SPSS, Inc., Chicago, IL, USA). Data from three or more
independent experiments were collected and analyzed as the mean ±
standard error of the mean. The significance of the results was
assessed using a paired t-test between two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Smoke exposure results in the
upregulation of IL1β
Evidence has been accumulating, which indicates that
the progression of liver disease is associated with cigarette
smoking (25). To assess the
inflammatory status in the liver upon smoking exposure, the present
study used a smoking-exposed rat model. Examination of liver
tissues by HE staining showed no significant structural alterations
or inflammatory infiltrates in the rats exposed to smoke for 14
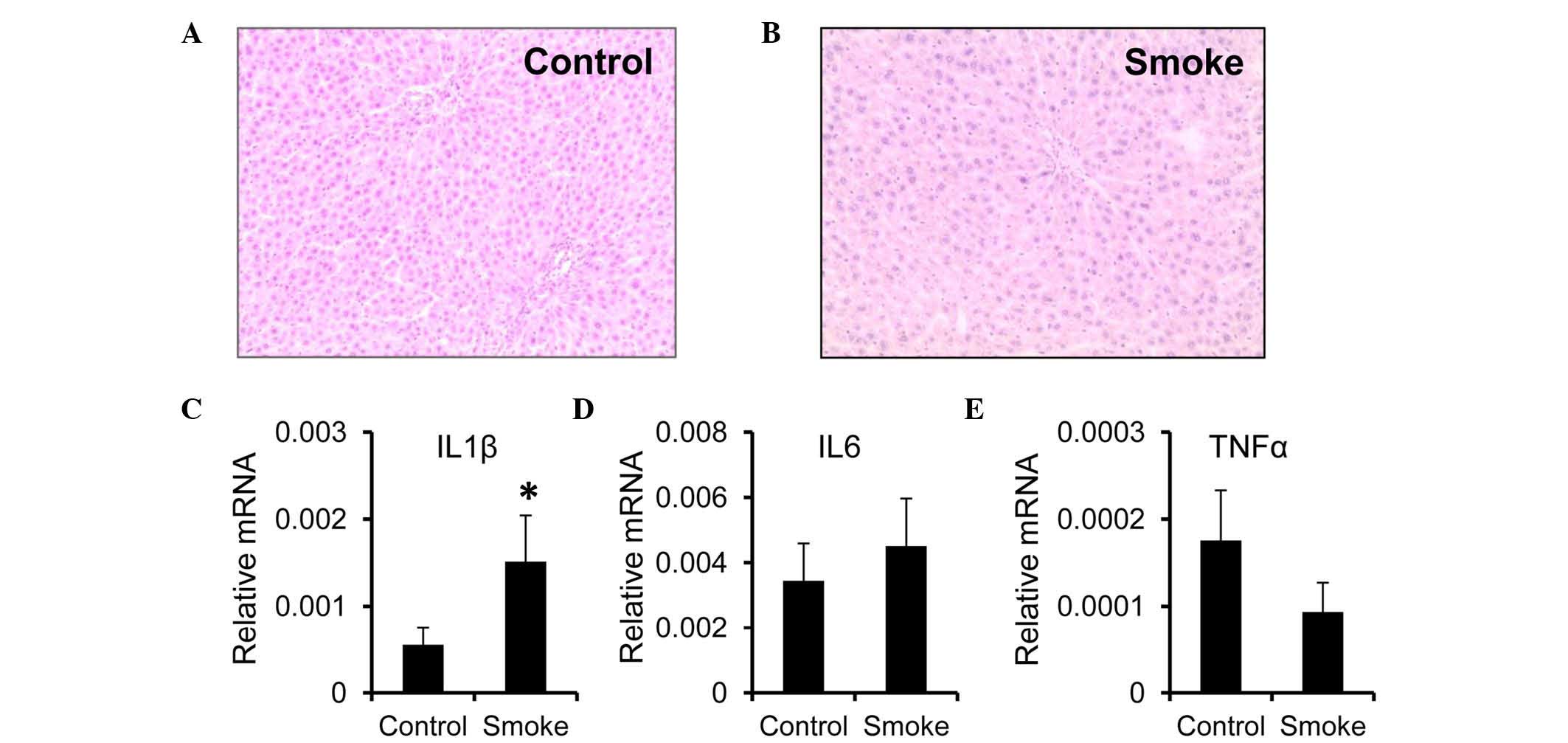
weeks (smoke group), compared with the control group (Fig. 1A and B). However, the mRNA
expression of IL1β was higher in the livers from the smoke group,
compared with those from the control group (Fig. 1C). Unlike IL1β, the mRNA expression
levels of IL6 and TNFα remained unchanged following smoke exposure
(Fig. 1D and E).
Inflammation is shown to involve cross-talk with
autophagy in the liver (23). To
evaluate the effects of smoking on autophagic activity in the
liver, RT-qPCR analysis was performed. The results revealed that
the expression of mammalian target of rapamycin (mTOR), an
inhibitor of autophagy, was decreased in the smoke group, compared
with the control group, although this was not significant (data not
shown). However, the expression of AMP-activated protein kinase
(Ampk), a negative regulator of the mTOR pathway, was elevated when
the rats were exposed to smoke (Fig.
2A). In addition, the expression of Wip1, a positive regulator
of the mTOR pathway, was decreased in the smoke group, compared
with the control group (Fig. 2B).
These findings indicated that autophagy in the liver may be induced
by smoking. To confirm this, the present study examined the effects
of smoking on the expression of autophagy-associated genes in the
liver tissues. The expression levels of Atg5 and Atg12 were
comparable between the smoke group and control group (Fig. 2C and D). However, the expression
levels of Ulk1 and annexin A3 (Anx3), a homolog of human
microtubule associated protein 1 light chain 3 (LC3) were
significantly higher in the smoke group, compared with those in the
control group (Fig. 2E and F).
Taken together, these data suggested that autophagy was upregulated
in the liver upon smoking exposure.
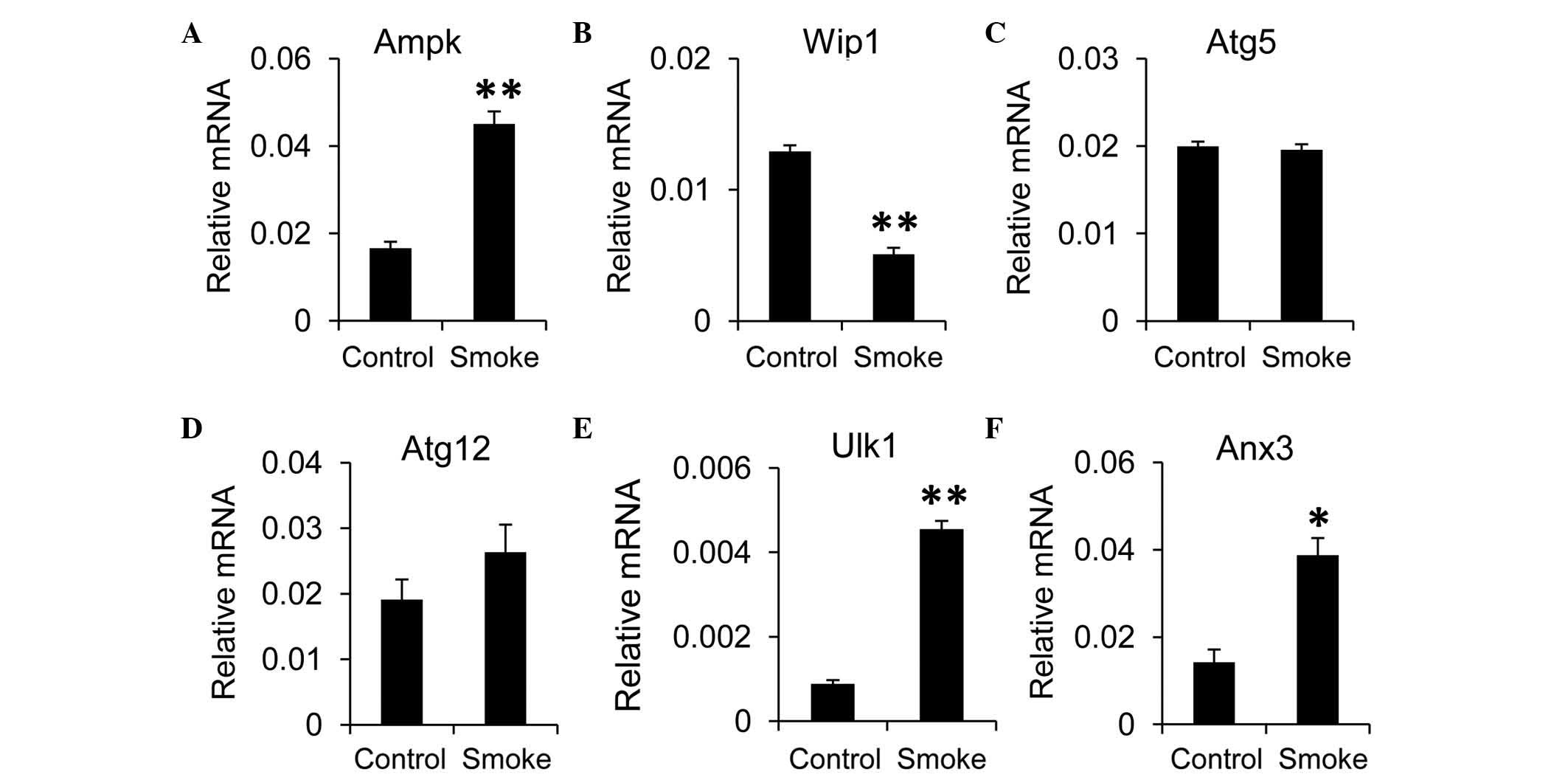 | Figure 2.Smoke exposure increases autophagy in
the liver. mRNA expression levels of the autophagy regulators, (A)
Ampk and (B) Wip1, and autophagy-associated components, including
(C) Atg5, (D) Atg12, (E) Ulk1 and (F) Anx3, were measured in the
liver using reverse transcription-quantitative polymerase chain
reaction analysis. Gapdh was used as the housekeeping gene. Data,
obtained from 10 rats per group, are presented as the mean ±
standard error of the mean. *P<0.05 and **P<0.01, compared
with the control group. Ampk, AMP-activated protein kinase; Wip1,
wild-type p53-induced phosphatase 1; Atg, autophagy-related
protein; Ulk1, unc-51-like autophagy activating kinase 1; Anx3,
annexin A3. |
Smoking induces a reduction in the
expression of rat hepatic Cyp genes
Cyp enzymes possess the capacity to catalyze the
oxidative biotransformation of the majority of drugs. To examine
whether smoking-induced inflammation alters hepatic function by
regulating the expression of Cyp, the present study measured the
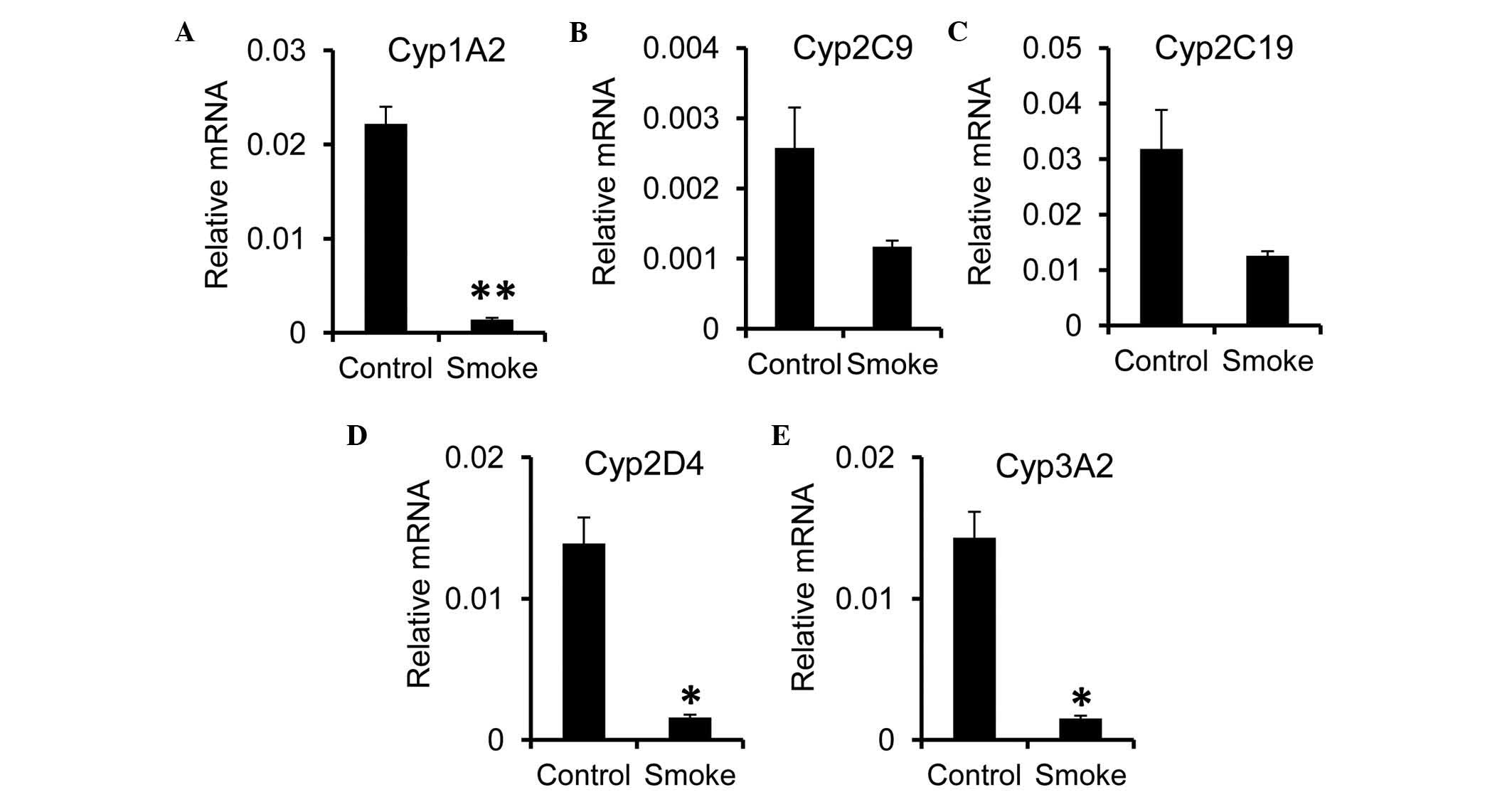
expression of Cyp genes in the liver. A significant reduction in
the mRNA expression level of Cyp1A2 was observed in the smoke
group, compared with the control group (Fig. 3A). For the Cyp2 family, the
expression of Cyp2D4, but not Cyp2C9 or Cyp2C19, was significantly
reduced following smoke exposure (Fig.
3B-D). As for the Cyp3 family, the expression of Cyp3A2 was
decreased in the smoke group (Fig.
3E). In concordance with previous reports showing a reduction
in the expression of Cyp during inflammation (26,27),
these data suggested that smoking-induced inflammation altered the
catalyzing capacity of the liver.
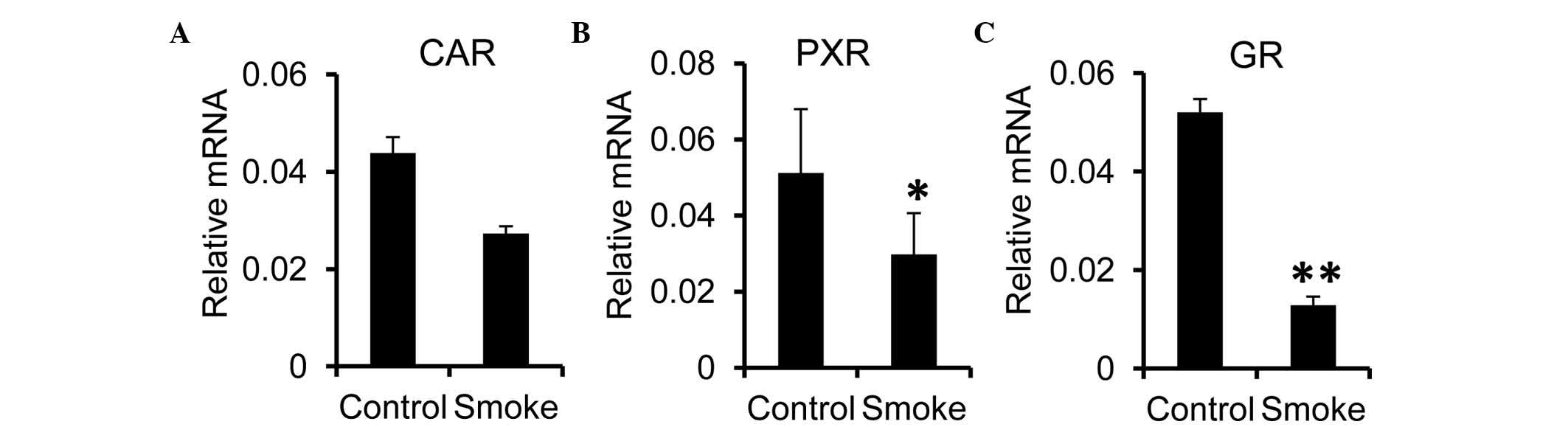
It is known that the gene expression of Cyp is
regulated by nuclear receptors, including PXR and CAR (28–30).
The present study investigated effects of smoking exposure on the
expression of these nuclear receptors in the liver. It was found
that the hepatic mRNA expression of CAR was comparable between the
control and smoke groups (Fig.
4A). However, the expression levels of PXR and GR were
significantly reduced in the smoke group, compared with the control
group (Fig. 4B and C). These
results suggested that the reduction in the hepatic expression of
Cyp in the smoking-exposed rats may have been attributed to the
reduced synthesis of nuclear receptors, including GR, in the
liver.
GR mediates the expression of CYP in
human hepatocytes
The present study used the LO2 human hepatocyte cell
line to evaluate the role of GR in regulating the expression of
CYP. The LO2 cells were treated with Dex alone or with Dex and the
GR inhibitor, RU486 for 2 days. LO2 cells are sensitive to Dex.
Compared with the control cells (Fig.
5A), treatment with Dex alone resulted in morphological
alterations, for example the LO2 cells became longer and more
stretched (Fig. 5B). These
morphological changes were absent when the LO2 cells were treated
with Dex and RU486 (Fig. 5C). Dex
exhibited differential effects on the expression of CYP by LO2
cells. Treatment of the LO2 cells with Dex alone led to an increase
in the expression of CYP3A4, however, this increase was reduced
when the LO2 cells were incubated with Dex and RU486 (Fig. 5D). Similarly, Dex treatment
resulted in upregulation of thee expression of CYP2C19. The
expression of CYP2C19 was decreased, although not significantly, in
cells treated with Dex and RU486, compared with the Dex group
(Fig. 5E). By contrast, treatment
with Dex alone resulted in a significant reduction in the
expression of CYP2C9 in LO2 cells (Fig. 5F). The expression of CYP2C9 in the
Dex and RU486 group returned to a level, which was comparable with
that of the control cells. However, Dex had no effect on the
expression of CYP2D6 (Fig. 5G).
Similar to CYP3A4, LO2 cells with Dex alone led to an increase in
the expression of CYP1A2, however, this increase was reduced when
the LO2 cells were incubated with Dex and RU486 (Fig. 5H). Collectively, these data
suggested that GR exhibited a differential role in regulating
hepatic CYP expression.
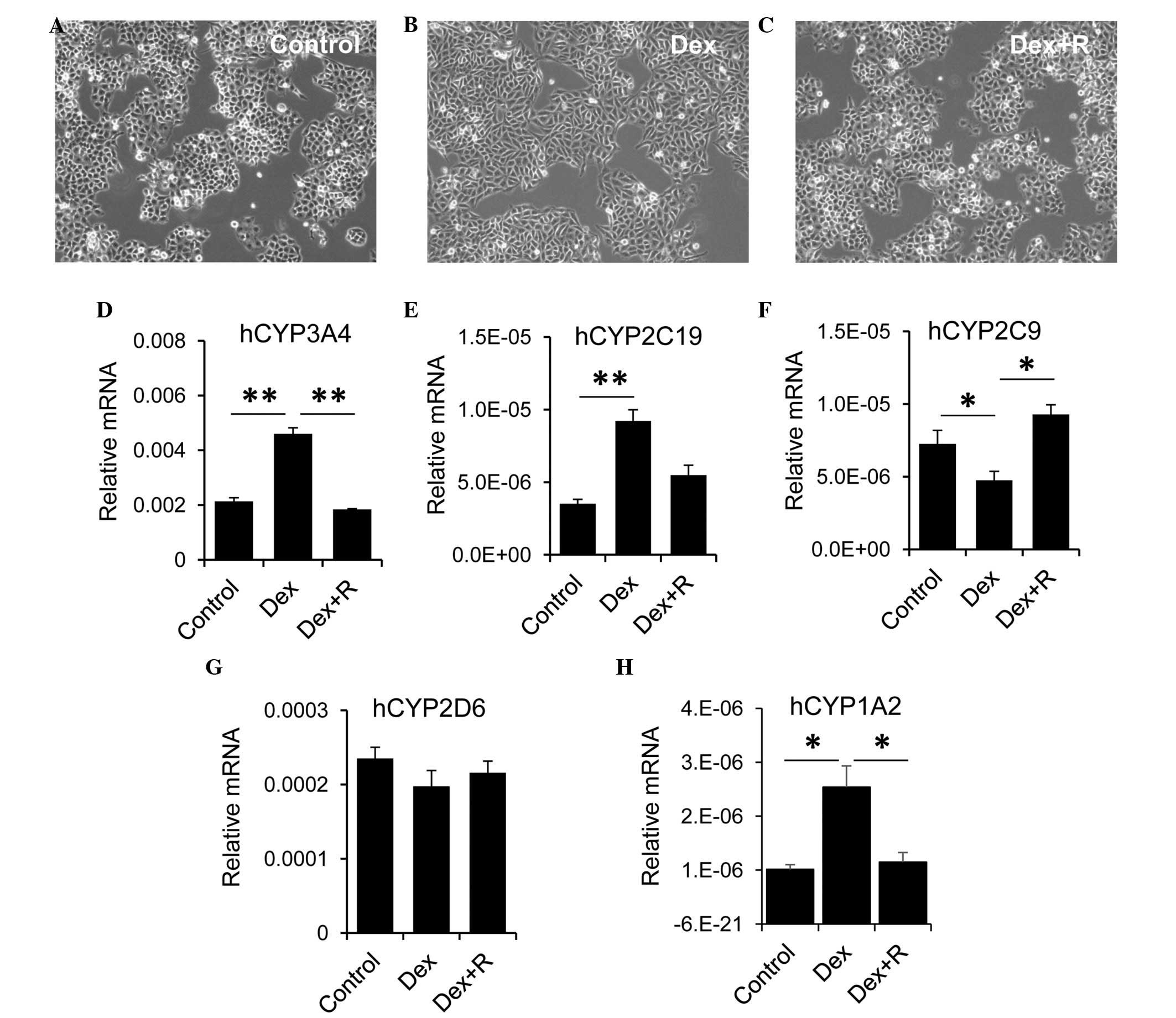 | Figure 5.GR is involved in regulating the
hepatic expression of CYP. Human liver LO2 cells were either (A)
untreated, treated with (B) Dex or treated with (C) Dex + R
(magnification, ×200). Following treatment, the LO2 cells were
harvested to measure the mRNA expression levels of (D) hCYP3A4, (E)
hCYP2C19, (F) hCYP2C9, (G) hCYP2D6 and (H) hCYP1A2. hGADPH was used
as the housekeeping gene. The data are presented as the mean +
standard error of the mean. *P<0.05 and **P<0.01, compared
with the control group. GR, glucocorticoid receptor; CYP,
cytochrome P450; Dex, dexamethasone; R, RU486. |
To evaluate effects of GR on autophagic activity in
human heptocytes, the present study measured autophagic genes in
LO2 cells in response to Dex. No significant change in the RNA
expression levels of mTOR, ATG5, ATG12, LC3 or ULK1 were found in
the Dex-treated cells, compared with the control cells or the Dex
and RU486-treated cells (Fig.
6A-E). Among the genes selected in the present study, only the
expression of Beclin 1 was reduced in the Dex-treated cells,
however, the addition of RU486 did not recover the expression level
(Fig. 6F). Taken together, these
data suggested that GR had minimal effect on the regulation of
autophagy in LO2 cells.
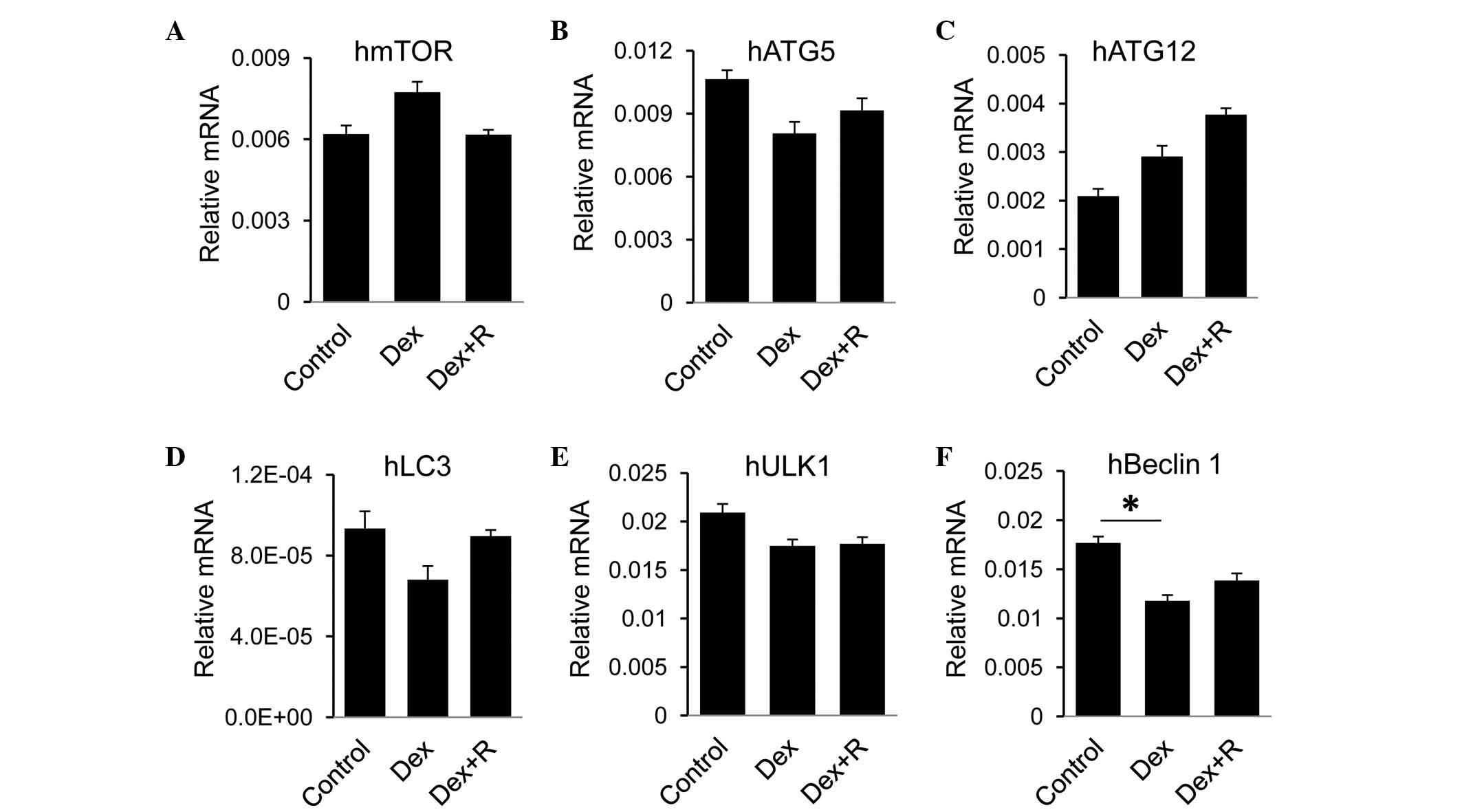 | Figure 6.GR has minimal effect on the hepatic
expression of CYP. Following treatment with Dex or Dex+R, the LO2
cells were harvested to measure the m RNA expression levels of the
autophagy-associated genes, (A) hmTOR, (B) hATG5, (C) hATG12, (D)
hLC3, (E) hULK1 and (F) hBeclin 1. hGADPH was used as the
housekeeping gene. Data are presented as the mean + standard error
of the mean. *P<0.05, compared with the control group. GR,
glucocorticoid receptor; CYP, cytochrome P450; Dex, dexamethasone;
R, RU486; mTOR, mammalian target of rapamycin; ATG,
autophagy-related protein; LC3, microtubule-associated protein 1
light chain 3; ULK1, unc-51-like autophagy activating kinase 1. |
Discussion
Cigarette smoking is considered to be a major
preventable contributor to morbidity and mortality rates worldwide.
In addition to the lungs, the liver is also affected by chemicals
produced from cigarettes, either directly or indirectly. In the
present study, a reduction in the expression levels of CYPs were
observed in the livers from cigarette smoking-exposed rats. The
data also suggested that autophagy was activated on exposure to
cigarette smoking. Using human liver LO2 cells, it was shown that
GR, at least in part, mediated the smoking-induced alteration in
the expression of CYPs.
CYPs constitute the major enzyme family capable of
catalyzing the oxidative biotransformation of the majority of
drugs, and are, therefore, of particular relevance for clinical
pharmacology (31,32). Among these enzymes, the CYP1, CYP2
and CYP3 families are responsible for metabolizing the majority of
drugs (33). Theophylline
represents a routine drug used to treat wheezing, shortness of
breath and chest tightness, which are normally observed in patients
with chronic bronchitis or emphysema (11). The chemicals derived from cigarette
smoke affect the lung predominantly by triggering airway
inflammatory responses, resulting in the development of emphysema
and respiratory bronchiolitis (34). In the present study, it was found
that the hepatic expression of Cyp1A2, which metabolizes
theophylline, was reduced in the smoking-exposed rats. Hussain
et al (35) reported that
incense smoke induces the expression of CYP1A2 (35). This discrepancy may be associated
with the severity of injury induced by the smoke. However, when
theophylline is used to treat patients with chronic bronchitis or
emphysema, its efficacy and retention may be altered in patients
who smoke. Notably, a significant reduction was also observed in
the expression of members of the CYP2 and CYP3 families. Therefore,
smoking cessation may be considered to provide optimal treatment
for patients who smoke.
It is known that CYPs are predominantly generated in
hepatocytes, the damage of which can affect the quantities and
qualities of CYPs. The severity of liver injury correlates
positively with levels of inflammation (36). Hepatic inflammation is prompted by
the binding of cyclic pro-inflammatory factors to toll-like
receptor 4 in hepatic Kupffer cells. The activated Kupffer cells
initiate the secretion of inflammatory cytokines, which in turn
activate NF-kB (37). The
nuclear-translocated NF-kB forms a complex with GR, prevents the
binding of GR to GR-responsive elements, and inhibits the
transcription of PXR and CAR (38,39).
The reduction in the levels of PXR and CAR, which bind to the
responsive elements of DNA, results in the inhibition of CYP3A
transcription and a subsequent decrease in its expression (40–42).
The role of GR in the regulation of CYPs has been a matter of
debate. Experiments using rats have supported the involvement of
this nuclear receptor in the regulation of CYPs. The present study
found that GR was decreased in the livers of rats exposed to smoke.
Using LO2 human liver cells, the present study showed that the
activation of GR with Dex upregulated the expression of CYP1A2,
CYP2C9 and CYP3A4, but downregulated the expression of CYP2C9. The
effect was reversed when the LO2 cells were treated with the Dex
and GR inhibitor, RU486. These data, together with previous
findings (43), demonstrated that
the GR signal exhibits a differential role in regulating the
hepatic expression of CYPs.
Autophagy, a cellular self-protective process
predominantly involving the recycling of its own nonessential
organelles to maintain homeostasis, can be activated by certain
stimuli (44). In the present
study, it was observed that autophagy was upregulated in the livers
of rats upon smoking exposure. However, smoking-induced autophagy
in the liver was independent of the GR signal. In certain cases,
autophagy or autophagy-associated proteins may induce apoptosis or
necrosis, and autophagy has been shown to degrade the cytoplasm
excessively, leading to autophagic cell death (45). Therefore, whether or not autophagy
is involved in hepatic metabolism through regulating the expression
of CYPs requires investigation addressed in the future.
In conclusion, the present study demonstrated an
association between cigarette smoking and metabolic alterations in
the liver by investigating the expression of CYP genes at the
transcriptional level. Future investigations on protein expression
levels are required to confirm these preliminary findings.
Experiments can be designed to treat emphysema in rats with
anti-inflammatory drugs. The hepatic metabolism of such drugs may
be evaluated and compared between smoking and control groups.
Another smoking cessation group can be included to evaluate the
contribution of smoking cessation to the hepatic metabolism of
drugs. These investigations are likely to provide clues to improve
treatment in patients who smoke.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 31471121 and 81270144) and
the Natural Science Foundation of Tianjin City (grant nos.
13JCYBJC22400, 13JCYBJC40000 and 14JCYBJC25700).
References
|
1
|
Kleinstreuer C and Feng Y: Lung deposition
analyses of inhaled toxic aerosols in conventional and less harmful
cigarette smoke: a review. International journal of environmental
research and public health. 10:4454–4485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Altamirano J and Bataller R: Cigarette
smoking and chronic liver diseases. Gut. 59:1159–1162. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bataller R: Time to ban smoking in
patients with chronic liver diseases. Hepatology. 44:1394–1396.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alebiosu CO: An update on ‘progression
promoters’ in renal diseases. J Natl Med Assoc. 95:30–42.
2003.PubMed/NCBI
|
|
5
|
Malfertheiner P and Schütte K: Smoking-a
trigger for chronic inflammation and cancer development in the
pancreas. Am J Gastroenterol. 101:160–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gershwin ME, Selmi C, Worman HJ, Gold EB,
Watnik M, Utts J, Lindor KD, Kaplan MM and Vierling JM: USA PBC
Epidemiology Group: Risk factors and comorbidities in primary
biliary cirrhosis: A controlled interview-based study of 1032
patients. Hepatology. 42:1194–1202. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mallat A, Hezode C and Lotersztajn S:
Environmental factors as disease accelerators during chronic
hepatitis C. J Hepatol. 48:657–665. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Azzalini L, Ferrer E, Ramalho LN, Moreno
M, Domínguez M, Colmenero J, Peinado VI, Barberà JA, Arroyo V,
Ginès P, et al: Cigarette smoking exacerbates nonalcoholic fatty
liver disease in obese rats. Hepatology. 51:1567–1576. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koh WP, Robien K, Wang R, Govindarajan S,
Yuan JM and Yu MC: Smoking as an independent risk factor for
hepatocellular carcinoma: The Singapore Chinese health study. Br J
Cancer. 105:1430–1435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zordoky BN, Aboutabl ME and El-Kadi AO:
Modulation of cytochrome P450 gene expression and arachidonic acid
metabolism during isoproterenol-induced cardiac hypertrophy in
rats. Drug Metab Dispos. 36:2277–2286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Danielson PB: The cytochrome P450
superfamily: Biochemistry, evolution and drug metabolism in humans.
Curr Drug Metab. 3:561–597. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guengerich FP: Cytochrome P450s and other
enzymes in drug metabolism and toxicity. AAPS J. 8:E101–E111. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martínez-Jimúnez CP, Jover R, Donato MT,
Castell JV and Gómez-Lechón MJ: Transcriptional regulation and
expression of CYP3A4 in hepatocytes. Curr Drug Metab. 8:185–194.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bell JC and Strobel HW: Regulation of
cytochrome P450 4F11 by nuclear transcription factor-kappaB. Drug
metabolism and disposition: the biological fate of chemicals.
40:205–211. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nannelli A, Chirulli V, Longo V and
Gervasi PG: Expression and induction by rifampicin of CAR- and
PXR-regulated CYP2B and CYP3A in liver, kidney and airways of pig.
Toxicology. 252:105–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Goodwin B, Hodgson E, D'Costa DJ,
Robertson GR and Liddle C: Transcriptional regulation of the human
CYP3A4 gene by the constitutive androstane receptor. Mol Pharmacol.
62:359–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pascussi JM, Gerbal-Chaloin S, Drocourt L,
Maurel P and Vilarem MJ: The expression of CYP2B6, CYP2C9 and
CYP3A4 genes: A tangle of networks of nuclear and steroid
receptors. Biochim Biophys Acta. 1619:243–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gerbal-Chaloin S, Daujat M, Pascussi JM,
Pichard-Garcia L, Vilarem MJ and Maurel P: Transcriptional
regulation of CYP2C9 gene. Role of glucocorticoid receptor and
constitutive androstane receptor. J Biol Chem. 277:209–217. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cooper BW, Cho TM, Thompson PM and Wallace
AD: Phthalate induction of CYP3A4 is dependent on glucocorticoid
regulation of PXR expression. Toxicol Sci. 103:268–277. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moszczyński P, Zabiński Z, Moszczyński P
Jr, Rutowski J, Słowinski S and Tabarowski Z: Immunological
findings in cigarette smokers. Toxicol Lett. 118:121–127. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ryter SW and Choi AM: Autophagy in the
lung. Proc Am Thorac Soc. 7:13–21. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Czaja MJ, Ding WX, Donohue TM Jr, Friedman
SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et
al: Functions of autophagy in normal and diseased liver. Autophagy.
9:1131–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo R, Xu X, Babcock SA, Zhang Y and Ren
J: Aldehyde dedydrogenase-2 plays a beneficial role in ameliorating
chronic alcohol-induced hepatic steatosis and inflammation through
regulation of autophagy. J Hepatol. 62:647–656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li H, Wu Q, Xu L, Li X, Duan J, Zhan J,
Feng J, Sun X and Chen H: Increased oxidative stress and disrupted
small intestinal tight junctions in cigarette smoke-exposed rats.
Mol Med Rep. 11:4639–4644. 2015.PubMed/NCBI
|
|
25
|
Corpechot C, Gaouar F, Chrétien Y, Johanet
C, Chazouillères O and Poupon R: Smoking as an independent risk
factor of liver fibrosis in primary biliary cirrhosis. J Hepatol.
56:218–224. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Slaviero KA, Clarke SJ and Rivory LP:
Inflammatory response: An unrecognised source of variability in the
pharmacokinetics and pharmacodynamics of cancer chemotherapy.
Lancet Oncol. 4:224–232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Aitken AE, Richardson TA and Morgan ET:
Regulation of drug-metabolizing enzymes and transporters in
inflammation. Annu Rev Pharmacol Toxicol. 46:123–149. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pondugula SR, Dong H and Chen T:
Phosphorylation and protein-protein interactions in PXR-mediated
CYP3A repression. Expert Opin Drug Metab Toxicol. 5:861–873. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chai XJ, Zeng S and Xie W: Nuclear
receptors PXR and CAR: Implications for drug metabolism regulation,
pharmacogenomics and beyond. Expert Opin Drug Metab Toxicol.
9:253–266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shah P, Guo T, Moore DD and Ghose R: Role
of constitutive androstane receptor in Toll-like receptor-mediated
regulation of gene expression of hepatic drug-metabolizing enzymes
and transporters. Drug Metab Dispos. 42:172–181. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Samer CF, Lorenzini KI, Rollason V, Daali
Y and Desmeules JA: Applications of CYP450 testing in the clinical
setting. Mol Diagn Ther. 17:165–184. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zanger UM and Schwab M: Cytochrome P450
enzymes in drug metabolism: Regulation of gene expression, enzyme
activities, and impact of genetic variation. Pharmacol Ther.
138:103–141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kelly SL and Kelly DE: Microbial
cytochromes P450: Biodiversity and biotechnology. Where do
cytochromes P450 come from, what do they do and what can they do
for us? Philos Trans R Soc Lond B Biol Sci. 368:201204762013.
|
|
34
|
Washko GR, Hunninghake GM, Fernandez IE,
Nishino M, Okajima Y, Yamashiro T, Ross JC, Estépar RS, Lynch DA,
Brehm JM, et al: Lung volumes and emphysema in smokers with
interstitial lung abnormalities. N Engl J Med. 364:897–906. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hussain T, Al-Attas OS, Al-Daghri NM,
Mohammed AA, De Rosas E, Ibrahim S, Vinodson B, Ansari MG and
El-Din KI: Induction of CYP1A1, CYP1A2, CYP1B1, increased oxidative
stress and inflammation in the lung and liver tissues of rats
exposed to incense smoke. Mol Cell Biochem. 391:127–136. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Minakata Y, Ueda H, Akamatsu K, Kanda M,
Yanagisawa S, Ichikawa T, Koarai A, Hirano T, Sugiura H, Matsunaga
K and Ichinose M: High COPD prevalence in patients with liver
disease. Intern Med. 49:2687–2691. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ivanenkov YA, Balakin KV and Lavrovsky Y:
Small molecule inhibitors of NF-kB and JAK/STAT signal transduction
pathways as promising anti-inflammatory therapeutics. Mini Rev Med
Chem. 11:55–78. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Assenat E, Gerbal-Chaloin S, Larrey D,
Saric J, Fabre JM, Maurel P, Vilarem MJ and Pascussi JM:
Interleukin 1beta inhibits CAR-induced expression of hepatic genes
involved in drug and bilirubin clearance. Hepatology. 40:951–960.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Widen C, Gustafsson JA and Wikström AC:
Cytosolic glucocorticoid receptor interaction with nuclear
factor-kappa B proteins in rat liver cells. Biochem J. 373:211–220.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beigneux AP, Moser AH, Shigenaga JK,
Grunfeld C and Feingold KR: Reduction in cytochrome P-450 enzyme
expression is associated with repression of CAR (constitutive
androstane receptor) and PXR (pregnane X receptor) in mouse liver
during the acute phase response. Biochem Biophys Res Commun.
293:145–149. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gu X, Ke S, Liu D, Sheng T, Thomas PE,
Rabson AB, Gallo MA, Xie W and Tian Y: Role of NF-kappaB in
regulation of PXR-mediated gene expression: A mechanism for the
suppression of cytochrome P-450 3A4 by proinflammatory agents. J
Biol Chem. 281:17882–17889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang YM, Ong SS, Chai SC and Chen T: Role
of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin
Drug Metab Toxicol. 8:803–817. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vrzal R, Stejskalova L, Monostory K,
Maurel P, Bachleda P, Pavek P and Dvorak Z: Dexamethasone controls
aryl hydrocarbon receptor (AhR)-mediated CYP1A1 and CYP1A2
expression and activity in primary cultures of human hepatocytes.
Chem Biol Interact. 179:288–296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Schneider JL and Cuervo AM: Liver
autophagy: Much more than just taking out the trash. Nat Rev
Gastroenterol Hepatol. 11:187–200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Marino G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014. View Article : Google Scholar : PubMed/NCBI
|