Introduction
Liver fibrosis is a common pathological process
occurring as a result of a variety of liver diseases and lesions.
Transforming growth factor (TGF)-β1 is an important promoter of
liver fibrosis (1), which
activates the differentiation of fibroblasts into myofibroblasts
and stimulates the latter to produce a large quantity of matrix
proteins (2).
In mammals, the TGF-β superfamily is composed of
three homologous genes, which may interact with each other: TGF-β1,
TGF-β2 and TGF-β3. Our previous study demonstrated that TGF-β3 may
significantly inhibit the expression of TGF-β1 and collagen I, the
primary protein of the extracellular matrix (3). A further study demonstrated that the
TGF-β3 promoter contains the cyclic adenosine monophosphate
(cAMP)-responsive element (CRE) site (4). CRE binding protein 1 (CREB-1),
phosphorylated (p) on Ser-133, combines with this particular site
and activates the TGF-β3 promoter, inducing the secretion of
TGF-β3. Therefore, p-CREB-1 is a critical transcription factor in
mediating TGF-β3 auto-induction (4).
Previous studies have indicated that p-CREB-1 has an
inhibitory effect on myocardial and pulmonary fibroses (5,6). In
addition, p-CREB-1 may have an antifibrolytic effect in liver.
However, the antifibrolytic effect is counteracted by the
inactivation of p-CREB-1 through dephosphorylation (7).
Post-translational modifications, including
phosphorylation, are the chemical modifications of
newly-synthesized polypeptide chains or proteins, which alter their
physical and chemical properties and therefore their biological
function. Certain studies have demonstrated that acetylation,
another post-translational modification, may alter the conformation
of a protein and result in a greater biological effect (8). The present study aimed to identify
the underlying mechanisms involved in post-translational
modifications of CREB-1 in inhibiting TGF-β1-mediated liver
fibrosis. The results indicated a theoretical basis for clarifying
the underlying CREB-1 antifibrolytic mechanisms, and thereby
provide a novel therapeutic target for the treatment of liver
fibrosis.
Materials and methods
Cell culture and reagents
HSC-T6 rat hepatic stellate cells (HSCs) were
provided by the Division of Gastroenterology Laboratory of Union
Hospital (Wuhan, China). HSCs were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS;
Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere of 5% CO2. All experiments were conducted
when cells were at an exponential stage of growth. Human
recombinant TGF-β1 was purchased from PeproTech, Inc. (Rocky Hill,
NJ, USA) and used at a concentration of 10 ng/ml. The
cAMP-elevating agent, forskolin (FSK) was purchased from Beyotime
Institute of Biotechnology (Haimen, China) and the histone
deacetylase (HDAC) inhibitor, trichostatin A (TSA) was obtained
from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
Transfection and treatment
HSCs were seeded into 6-well plates at a
concentration of 2×105/well. Transfection with
pRSV-CREB-1 expression vector or an empty vector (Invitrogen;
Thermo Fisher Scientific, Inc.) was conducted when cells reached
70–90% confluence. Lipofectamine™ 2000 (10 µl; Invitrogen; Thermo
Fisher Scientific, Inc.) in 250 µl Opti-minimal essential medium
(Opti-MEM; Gibco; Thermo Fisher Scientific, Inc.) was mixed with 4
µg vector in 250 µl Opti-MEM and incubated for 20 min at room
temperature to allow formation of the transfection complex, which
was then added to each well (500 µl transfection complex in 1,500
µl DMEM). A total of 6 h following transfection, culture media were
replaced with fresh DMEM, and cells were incubated for an
additional 18 h. TGF-β1, FSK or TSA were subsequently added into
different wells, to a final concentration of 10 ng/ml, 10 µM and
100 nM respectively. Treated HSCs were incubated for various times
and harvested for subsequent experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (Ambion; Thermo Fisher Scientific, Inc.). The purity and
concentration of the extracted RNA was detected by an ultraviolet
spectrophotometer (Eppendorf Biophotometer Plus; Thermo Fisher
Scientific, Inc.), and reverse-transcribed to cDNA using
PrimeScript™ RTase kit (Takara Biotechnology Co., Ltd., Dalian,
China) according to the manufacturer's protocol. qPCR was performed
in a total volume of 10 µl with 5 µl SYBR Green PCR Master mix
(Takara Biotechnology Co., Ltd.), 0.5 µl of each primer (0.2 µmol
in total), 1 µl cDNA (1:10 dilution) and 3 µl double-distilled
water. Primer sequences are presented in Table I. All cDNA samples were tested in
duplicate using a StepOne™ PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.), with thermocycling conditions as follows:
An initial denaturation step of 50°C for 2 min and 95°C for 10 min,
followed by 40 cycles of denaturation at 95°C for 15 sec and
annealing at 60°C for 1 min. Fluorescence data from each sample was
analyzed using the 2−ΔΔCq method (9), with GAPDH gene expression serving as
an endogenous control.
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction.
|
| Sequence (5′-3′) |
|---|
|
|
|
|---|
| Gene | Sense | Antisense |
|---|
| CREB-1 |
CAGTGCCAACCCCGATTTA |
TTGCTCCTCCCTGGGTAATG |
| Collagen I |
TTGTGCGATGACGTGATCTGT |
TTGGTCGGTGGGTGACTCTG |
| Smad3 |
GGGCCTGCTGTCCAATGT |
AATGTGCCGCCTTGTAAGCT |
| Smad7 |
TGGATGGCGTGTGGGTTTA |
TGGCGGACTTGATGAAGATG |
| ERK1/2 |
TTTGGTCTGTGGGCTGCAT |
TCCTGGGAAGATAGGCCTGTT |
| RhoA |
TCGGAATGATGAGCACACAAG |
GGTTTTACCGGCTCCTGCTT |
| ROCK1 |
TTCATTCCTACCCTCTACCACT |
GGCTTAAAAACATGCCACAA |
| GAPDH |
GTATGACTCTACCCACGGCAAGT |
TTCCCGTTGATGACCAGCTT |
Extraction of nuclear protein
Nuclear proteins from HSCs cultured in 100-mm
culture dishes were extracted using a Nuclear-Cytosol Extraction
kit (Applygen Technologies, Inc., Beijing, China). Cells were
washed with cold PBS three times, lysed in 300 µl cytoplasm
extraction buffer A for 10 min and 20–30 µl cytoplasm extraction
buffer B for 1 min. Following a 5 min centrifugation at 1,000 × g,
the pellet containing nuclei was collected and resuspended in
70–100 µl nuclear extraction buffer for 30 min. The solution was
centrifuged at 12,000 × g for 10 min, and the supernatant
containing nuclear proteins was collected and stored at −70°C until
analysis. All procedures were carried out at 4°C. Protein
concentrations were determined using a Bicinchoninic Acid Protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.).
Western blotting
Whole-cell lysates were obtained using
radioimmunoprecipitation buffer (Applygen Technologies, Inc.)
containing protein phosphatase inhibitor (Applygen Technologies,
Inc.), according to the manufacturers' protocol; the nuclear
extracts were obtained as described above. Equal quantities of
protein (30–50 µg) from the lysates were separated by 10% SDS-PAGE
and transferred to polyvinylidene difluoride membranes. Membranes
were blocked with 5% non-fat dry milk in TBS containing 0.1%
Tween-20 (TBST) for 1 h at room temperature, and incubated
individually with the following primary antibodies: Rabbit
anti-CREB-1 (1:1,000; catalog no. 9197; Cell Signaling Technology,
Inc., Danvers, MA, USA), rabbit anti-mothers against
decapentaplegic (Smad) 3 (1:1,500; catalog no. 1735-1; Epitomics,
Burlingame, CA, USA), rabbit anti-extracellular signal-regulated
kinase 1/2 (ERK1/2; 1:1,000; catalog no. 4695; Cell Signaling
Technology, Inc.), rabbit anti-collagen I (1:5,000; catalog no.
ab34710; Abcam, Cambridge, UK) and rabbit anti-GAPDH (1:1,000;
catalog no. GTX100118; GeneTex, Inc., Irvine, CA, USA), overnight
at 4°C. Separate membranes were blocked with 5% bovine serum
albumin (MP Biomedicals, Santa Ana, CA, USA) in TBST for 1 h at
room temperature, and incubated with rabbit anti-p-CREB (Ser-133;
1:1,000; catalog no. 9198; Cell Signaling Technology, Inc.), goat
anti-p-Smad2/3 (Ser-423/425; 1:100; catalog no. sc-11769; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) and rabbit anti-p-ERK1/2
(Thr-202/Tyr-204; 1:1,000; catalog no. 4370S; Cell Signaling
Technology, Inc.). Following three washes in TBST, membranes were
incubated with anti-rabbit or anti-goat IgG horseradish
peroxidase-conjugated secondary antibodies (1:8,000; catalog nos.
111-035-003 and 305-035-003; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA) for 1 h at room temperature. Target
proteins were detected using an Enhanced Chemiluminescence
Detection kit (Thermo Fisher Scientific, Inc.). Densitometric
analysis was performed using AlphaView Stand Alone software version
3.4.0 (ProteinSimple, San Jose, CA, USA).
Statistical analysis
All experiments were performed three times
independently. Data were analyzed using SPSS software version 17.0
(SPSS, Inc., Chicago, IL, USA), using unpaired Student's
t-tests and one-way analyses of variance followed by
Fisher's least significant difference post-hoc test. Data are
presented as the mean ± standard deviation. P<0.05 was
considered to indicate a statistically significant difference.
Graphs were produced using GraphPad Prism software version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA).
Results
cAMP-elevating agent FSK activates
CREB-1
HSCs were stimulated with the cAMP-elevating agent,
FSK for various times. The protein expression levels of CREB-1
remained unaltered regardless of the duration of stimulation
(Fig. 1A). However,
phosphorylation levels of CREB-1 at Ser-133 peaked at 30 min
(1.9-fold greater than control group; P=0.011), then gradually
declined, and maintained a relatively stable level after 1 h
(Fig. 1A and B), demonstrating
inconsistencies with a previous study on cardiac fibroblasts
(10). In addition, CREB-1 mRNA
expression levels following FSK stimulation in HSCs were examined.
There were no statistically significant differences between the
groups at any time points (P=0.863; Fig. 1C). These results indicate that
CREB-1 is activated by FSK in a time-dependent manner.
CREB-1 decreases collagen I expression
in HSCs
A pRSV-CREB-1 expression vector and an empty vector
were used to transfect HSCs, to investigate whether CREB-1
inhibited collagen I expression. Transfection with pRSV-CREB-1
expression vector increased CREB-1 mRNA expression levels
(161.3-fold greater than the empty vector group; P<0.001;
Fig. 2A), which was accompanied by
a substantial increase in CREB-1 protein expression levels
(2.4-fold greater than the empty vector group; P=0.003; Fig. 2B and C) and a slight increase in
p-CREB-1 protein expression (P=0.056; Fig. 2D). HSCs were treated with FSK 24 h
following pRSV-CREB-1 transfection. Activated CREB-1 significantly
reduced the expression of collagen I with a peak at 0.5 h (1.5-fold
lower than the empty vector group; P=0.003), consistent with the
peak of CREB-1 activation. However, the negative effect of CREB-1
on collagen I protein expression levels gradually decreased or was
lost over time (Fig. 2E-H).
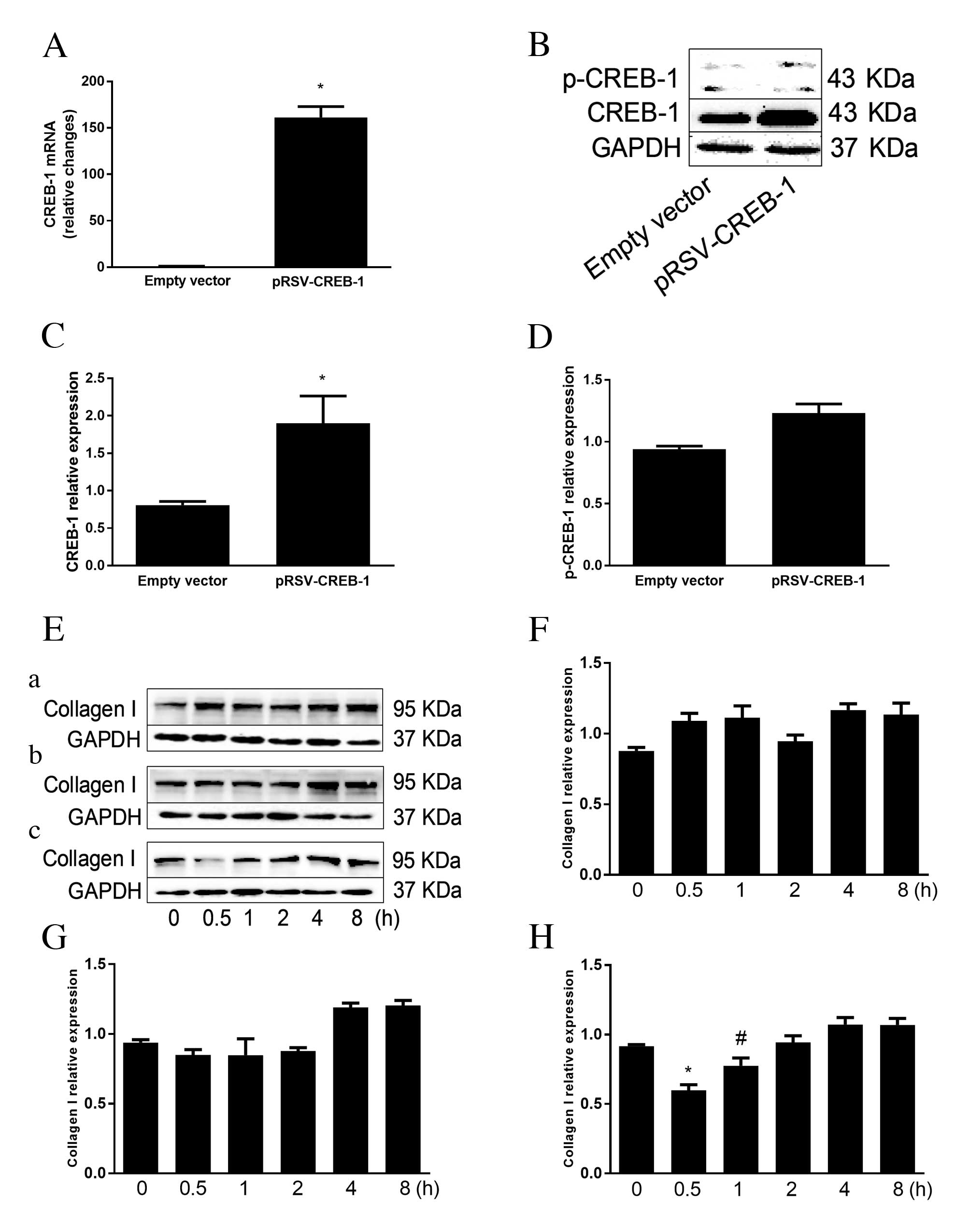 | Figure 2.CREB-1 decreases collagen I expression
in HSCs. (A-D) HSCs were transfected with pRSV-CREB-1 expression
vector or empty vector. A total of 24 h after transfection (A)
CREB-1 mRNA was detected by reverse transcription-quantitative
polymerase chain reaction and normalized to GAPDH expression, and
(B and C) CREB-1 and (D) p-CREB-1 nuclear protein expression levels
were detected by western blotting. Transfection with pRSV-CREB-1
expression vector increased CREB-1 mRNA and protein expression
levels, and slightly increased p-CREB-1 protein expression levels,
compared with the empty vector control. (E) HSCs were transfected
with (a) empty vector, (b) pRSV-CREB-1 expression vector or (c)
pRSV-CREB-1 expression vector, followed by treatment with 10 µM FSK
24 h after transfection. Total protein was extracted and collagen I
was detected by western blotting. Quantification of (F) empty
vector, (G) pRSV-CREB-1 expression vector and (H) pRSV-CREB-1
expression vector followed by FSK treatment. Activated CREB-1
significantly reduced the expression of collagen I with a peak at
0.5 h. The negative effect of CREB-1 on collagen I protein
expression levels gradually decreased or was lost over time. Data
are presented as the mean ± standard deviation of at least three
independent experiments. (A-D) *P<0.01 vs. empty vector group;
(E-H) #P<0.05 and *P<0.01 vs. 0 h group. CREB-1,
cyclic adenosine monophosphate-response element binding protein-1;
HSCs, hepatic stellate cells; p, phosphorylated; FSK,
forskolin. |
CREB-1 decreases collagen I expression
via inhibiting the Smad-dependent TGF-β1 signaling pathway
To elucidate the underlying mechanism involved in
CREB-1 inhibition of TGF-β1-mediated liver fibrosis, TGF-β1 and/or
FSK were used to treat transfected cells. As presented in Fig. 3A and B, TGF-β1 treatment
significantly increased collagen I protein expression levels in the
empty vector group (P=0.025); however, the protein expression
levels of collagen I demonstrated only a slight increase under
TGF-β1 stimulation in the pRSV-CREB-1 expression vector group. In
addition, the collagen I expression level was reduced in each
pRSV-CREB-1 group compared with the corresponding empty vector
group. The proteins of the Smad family were the first identified
substrates of type I receptor kinases and are key in the TGF-β1
signaling pathway. p-Smad2/3 is considered the final
phosphorylation step and the component of the p-Smad complex that
translocates to the nucleus to regulate target genes. p-Smad2/3
protein expression levels were consistent with collagen I; however,
p-Smad2/3 expression was suppressed significantly in the FSK plus
TGF-β1-treated group following CREB-1 overexpression (P<0.001;
Fig. 3C). No differences were
observed in Smad3 mRNA expression levels between groups (P=0.177;
Fig. 3D). The antagonistic Smad7
acts in opposition to p-Smads (11). In the present study, Smad7 mRNA
expression levels were greater in the FSK-treated group compared
with the TGF-β1-treated group (Fig.
3E). In addition, Smad7 mRNA expression levels were greater in
each pRSV-CREB-1 group, compared with the respective empty vector
control, particularly in the FSK plus TGF-β1-treated group
(P<0.001).
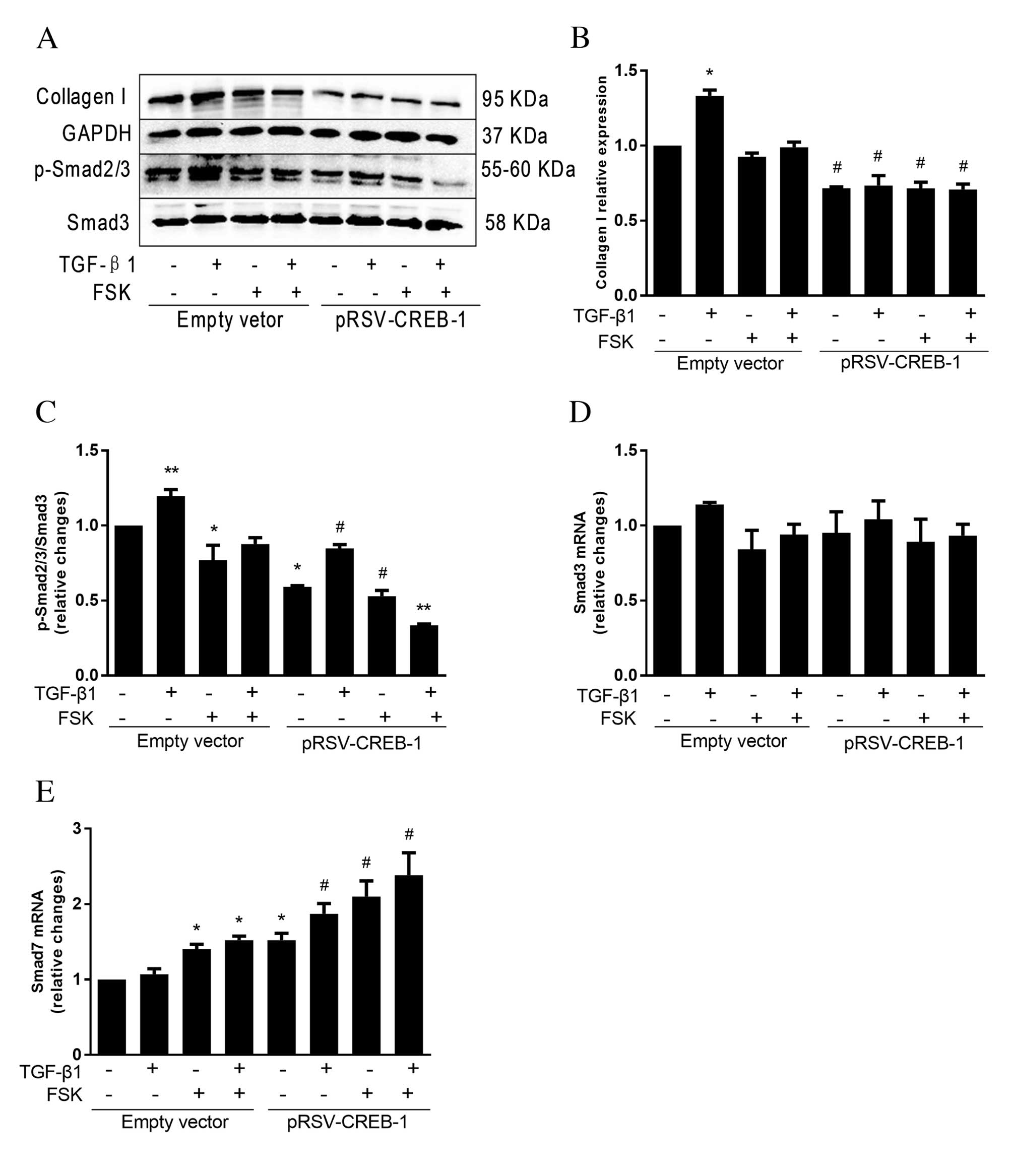 | Figure 3.CREB-1 inhibits the TGF-β1 signaling
pathway via Smad-dependent pathways. Cells were transfected with
pRSV-CREB-1 expression vector or empty vector, and 24 h later were
treated with 10 µM FSK and/or 10 ng/ml TGF-β1 for 1 h. (A)
Whole-cell lysates were analyzed by western blotting using
antibodies against collagen I, p-Smad2/3 (Ser-423/425), Smad3 or
GAPDH. (B) TGF-β1 significantly increased collagen I protein
expression levels in the empty vector group, but not in the
pRSV-CREB-1 expression vector group. (C) p-Smad2/3 protein
expression levels were consistent with collagen I; however,
p-Smad2/3 expression was suppressed in the FSK plus TGF-β1-treated
group following CREB-1 overexpression. Total RNA was collected and
Smad3 and Smad7 mRNA expression levels were detected by reverse
transcription-quantitative polymerase chain reaction. (D) No
differences were observed in Smad3 mRNA expression levels between
groups. (E) Smad7 mRNA levels were increased following transfection
with the pRSV-CREB-1 expression vector, compared with the empty
vector control. mRNA results were normalized to GAPDH expression.
Data are expressed as the mean ± standard deviation of at least
three independent experiments. *P<0.05 vs. no treatment group;
**P<0.05 vs. all other groups; #P<0.05 vs.
corresponding empty vector control group. CREB-1, cyclic adenosine
monophosphate-response element binding protein-1; TGF-β1,
transforming growth factor-β1; FSK, forskolin; Smad, mothers
against decapentaplegic; p, phosphorylated. |
CREB-1 decreases collagen I expression
via inhibition of the Smad-independent TGF-β1 signaling
pathway
p-ERK1/2 and Ras homolog gene family member A
(RhoA)/Rho-associated coiled-coil containing protein kinase 1
(ROCK1) signaling are involved in the TGF-β1 signaling pathway in
cardiac fibroblasts and corneal myofibroblasts (5,12).
These studies indicated that increased cAMP markedly blocks
TGF-β1-mediated activation of collagen synthesis and α-smooth
muscle actin expression via inhibition of ERK1/2 phosphorylation
and RhoA activation. The present study hypothesized that activated
CREB-1 has a similar role in HSCs. To verify this, p-ERK1/2 and
ERK1/2 protein expression levels were analyzed by western blotting,
and ERK1/2, RhoA and ROCK1 mRNA expression levels were analyzed via
RT-qPCR following CREB-1 overexpression. The results demonstrated
inconsistencies with collagen I and p-Smad2/3 data, as p-ERK1/2
levels were slightly, but not significantly, increased by FSK
stimulation alone following CREB-1 overexpression (P=0.201).
However, activated CREB-1 significantly attenuated activation of
ERK1/2 induced by TGF-β1 (P<0.001; Fig. 4A and B). No statistically
significant differences were observed in ERK1/2 mRNA expression
levels (P=0.453; Fig. 4C). TGF-β1
stimulated the expression of RhoA (Fig. 4D) and ROCK1 (Fig. 4E); however, overexpression of
CREB-1 reduced RhoA and ROCK1 mRNA expression levels, weakening the
effect of TGF-β1 stimulation.
 | Figure 4.CREB-1 inhibits the TGF-β1 signaling
pathway via Smad-independent pathways. Cells were transfected with
a pRSV-CREB-1 expression vector or an empty vector and 24 h later
treated with 10 µM FSK and/or 10 ng/ml TGF-β1 for 1 h. (A)
Whole-cell lysates were analyzed by western blotting using
antibodies against p-ERK1/2 (Thr-202/Tyr-204) and ERK1/2. (B)
Quantification of western blotting revealed that CREB-1
overexpression significantly attenuated the activation of ERK1/2
induced by TGF-β1. The total RNA was collected, and (C) ERK1/2, (D)
RhoA and (E) ROCK1 mRNA expression levels were detected by reverse
transcription-quantitative polymerase chain reaction. TGF-β1
stimulated the expression of RhoA and ROCK1; however, CREB-1
overexpression weakened the effect of TGF-β1 stimulation. mRNA
results were normalized to GAPDH expression. Data are expressed as
the mean ± standard deviation of at least three independent
experiments. *P<0.05 vs. no treatment group; **P<0.05 vs. all
other groups; #P<0.05 vs. corresponding empty vector
control group. ERK1/2, extracellular signal-regulated kinase 1/2;
CREB-1, cyclic adenosine monophosphate response element binding
protein-1; FSK, forskolin; TGF-β1, transforming growth factor-β1;
p, phosphorylated; RhoA, Ras homolog gene family member A; ROCK1,
Rho-associated coiled-coil containing protein kinase 1. |
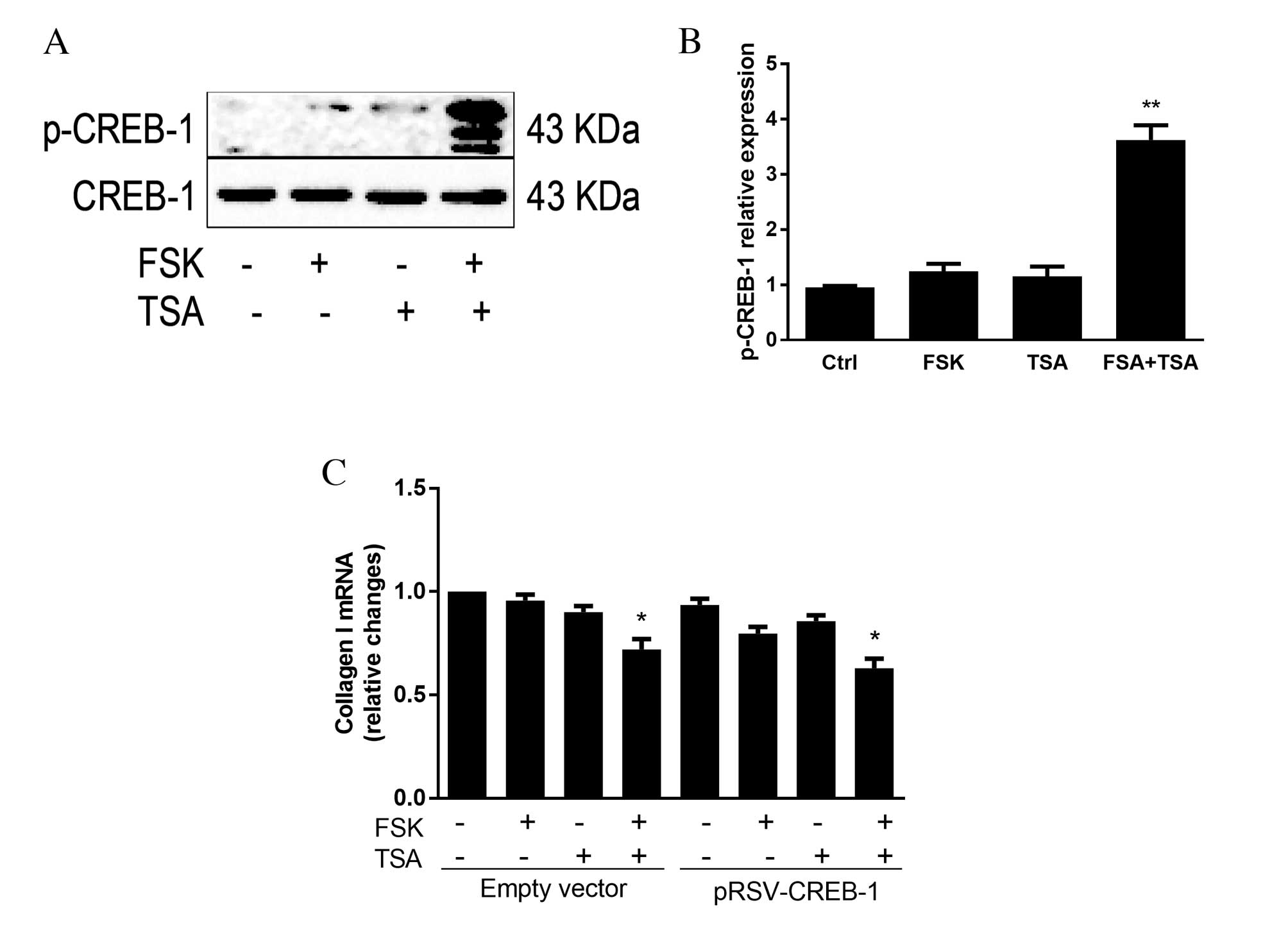
Acetylation extends the function of
phosphorylated CREB-1 in decreasing expression levels of collagen
I
The above results indicated that CREB-1 may be
activated by FSK, and have a key antifibrolytic role. However,
inactivated p-CREB-1 diminishes the effect of FSK stimulation. A
previous study revealed that the HDAC inhibitor TSA acetylates
CREB-1 and extends its phosphorylation level in an NIH/3T3 D5 cell
line (13). To determine whether
TSA has the same effect in HSCs, cells were stimulated with FSK
and/or TSA to induce CREB-1 post-translational modification. The
results demonstrated that in the FSK plus TSA group, protein
expression levels of p-CREB-1 were significantly increased compared
with the control group (3.6-fold greater than control group;
P<0.001; Fig. 5A and B).
Differences in CREB-1 expression between the other groups was not
statistically significant (P=0.139). In addition, following 4 h of
treatment with FSK and TSA in the pRSV-CREB-1 group, collagen I
mRNA expression levels were significantly reduced by 37% compared
with the no treatment group (P=0.011; Fig. 5C).
Discussion
As an important member of the activating
transcription factors/CREB family, CREB-1 is involved in a variety
of biological functions, including inhibiting inflammation and
apoptosis, enhancing immunity and memory, and regulating growth and
development (12,14). Previous studies have revealed that
that CREB-1 has antifibrolytic effects in lung, myocardial and
corneal tissue (5,6,15).
However, the role of CREB-1 in liver fibrosis remains to be
elucidated. The results of the present study demonstrated that
CREB-1 activated by FSK significantly reduces collagen I expression
in HSCs. This suggested that CREB-1 exerts an antifibrolytic effect
in liver tissue.
TGF-β1 has an important role in the liver fibrotic
response, and inhibition of the TGF-β1 signaling pathway is a
potential therapeutic strategy for the treatment of fibrosis.
TGF-β1 binds to TGF-β1 receptors, which induce the phosphorylation
of Smad2. Smad2 in turn phosphorylates the highly analogous
protein, Smad3, referred to as R-Smad, and induces the recruitment
of Smad4. The activated Smad3/4 complex translocates into the
nucleus to interact with Smad-binding elements and regulate
downstream genes, particularly collagen (16). By contrast, Smad7 acts in
opposition to this process by binding to the type I receptor and
preventing recruitment of phosphorylated Smad2 (17). TGF-β1 activates Smad proteins,
ERK1/2 and RhoA/ROCK1 (11,18,19).
In the present study, TGF-β1 induced the phosphorylation of Smad2/3
in HSCs, as well as the phosphorylation of ERK1/2 and the
expression of RhoA/ROCK1. This effect was significantly reduced
following treatment with FSK. In addition, activation of CREB-1
enhanced expression of the inhibitory factor, Smad7. Notably, the
combined treatment of TGF-β1 and FSK in CREB-1-transfected HSCs
resulted in a greater reduction in p-Smad2/3 and p-ERK1/2
expression levels and a greater increase in Smad7 expression levels
compared with FSK treatment alone. These results suggested that
activated CREB-1 has a strong inhibitory effect on TGF-β1-mediated
activation during fibrotic responses via blocking Smad-dependent
and -independent signaling pathways. However, other signaling
pathways, including phosphatidylinositol-3-kinase/serine-threonine
kinase, c-Jun N-terminal kinase, mitogen-activated protein kinase
and TGF-β-activated kinase 1 have been reported to be associated
with fibrosis (20–23). The specific mechanism underlying
CREB-1 regulation in HSCs remains to be fully elucidated.
Post-translational modifications of CREB-1 include
phosphorylation, acetylation, ubiquitination, sumoylation and
glycosylation (8). Phosphorylation
has an important role in transcriptional activation and biological
functions. Without phosphorylation, CREB-1 has almost no biological
activity. However, phosphorylated CREB-1 may be hydrolyzed in
internal environments. A previous study revealed that acetylation
may improve the stability of phosphorylated proteins by altering
the space conformation of CREB-1 and allowing its escape from
hydrolysis (13). Results from the
present study demonstrated that p-CREB-1 reached a peak level
following 30 min of FSK treatment, then declined gradually. The
previous study on cardiac fibroblasts demonstrated that the peak
level was reached at 5 min and then declined rapidly (10). This may have been due to the
experimental conditions, or the fact that the former was a cell
line, whereas the latter was a primary cell (10). HSCs treated with cAMP-elevating
agent or cAMP-elevating agent plus HDAC inhibitor for 4 h
demonstrated a significant increase in the expression of
Ser-133-phosphorylated CREB compared with FSK treatment alone.
Furthermore, co-treatment reduced collagen I mRNA expression levels
significantly in the CREB-1 overexpression group. These results
suggested that a HDAC inhibitor may prolong p-CREB-1
phosphorylation at Ser-133 and enhance its antifibrolytic
capacity.
CREB-binding protein (CBP) was originally identified
as a co-activator of CREB. CBP acts as a bridging factor between
CREB and transcription factor IIB. CBP, containing its homologue
p300, possesses intrinsic histone acetyltransferase activity;
CBP/p300 acetylates histones and non-histone proteins, including
transcription factors (24). The
phosphorylation of CREB at Ser-133 leads to the association of CREB
via the kinase-inducible domain with a short domain in CBP termed
the kinase-inducible domain interacting domain; p-CREB is further
acetylated, and CREB activity during the late attenuation phase is
prolonged (25). However, CBP, as
a broad-spectrum coactivator, directly interacts with R-Smad-Smad4
via the C-terminal domain to activate the transcription of
TGF-β1-responsive genes (26).
Therefore, CREB-1 may compete with R-Smad-Smad4 to interact with
CBP. In the present study, the expression of collagen I was
significantly reduced by FSK and TSA treatment following CREB-1
overexpression, demonstrating that excessive acetylated p-CREB-1
may inhibit R-Smad-Smad4 recruitment of CBP, thus inhibiting TGF-β1
signaling pathways. However, additional studies including in
vivo experiments are required to provide further evidence for
the antifibrolytic effect of acetylated p-CREB-1.
In conclusion, the results of the present study
demonstrated that p-CREB-1 inhibits TGF-β1-mediated liver fibrosis
by blocking the Smad-dependent and -independent signaling pathways
in HSCs. Furthermore, the results of the current study revealed
that CREB-1 is not sustainably activated; however, acetylation
extends CREB-1 phosphorylation and enhances its antifibrolytic
effect. The results of the present study suggested that CREB-1 may
be a useful potential therapeutic target for the treatment of liver
fibrosis.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81170401).
References
|
1
|
He Y, Huang C, Sun X, Long XR, Lv XW and
Li J: MicroRNA-146a modulates TGF-beta1-induced hepatic stellate
cell proliferation by targeting SMAD4. Cell Signal. 24:1923–1930.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van der Smissen A, Samsonov S, Hintze V,
Scharnweber D, Moeller S, Schnabelrauch M, Pisabarro MT and
Anderegg U: Artificial extracellular matrix composed of collagen I
and highly sulfated hyaluronan interferes with TGFβ(1) signaling
and prevents TGFβ(1)-induced myofibroblast differentiation. Acta
Biomater. 9:7775–7786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang Y, Liu P, Gao X, Qian W and Xu K:
rAAV2-TGF-β(3) decreases collagen synthesis and deposition in the
liver of experimental hepatic fibrosis rat. Dig Dis Sci.
55:2821–2830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deng L, Li Y, Huang JM, Zhou GY, Qian W
and Xu KS: Effects of p-CREB-1 on transforming growth factor-β3
auto-regulation in hepatic stellate cells. J Cell Biochem.
112:1046–1054. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chan EC, Dusting GJ, Guo N, Peshavariya
HM, Taylor CJ, Dilley R, Narumiya S and Jiang F: Prostacyclin
receptor suppresses cardiac fibrosis: Role of CREB phosphorylation.
J Mol Cell Cardiol. 49:176–185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu X, Sun SQ and Ostrom RS: Fibrotic lung
fibroblasts show blunted inhibition by cAMP due to deficient cAMP
response element-binding protein phosphorylation. J Pharmacol Exp
Ther. 315:678–687. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu X, Sun SQ, Hassid A and Ostrom RS:
cAMP Inhibits transforming growth factor-beta-stimulated collagen
synthesis via inhibition of extracellular signal-regulated kinase
1/2 and smad signaling in cardiac fibroblasts. Mol Pharmacol.
70:1992–2003. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johannessen M, Delghandi MP and Moens U:
What turns CREB on? Cell Signal. 16:1211–1227. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Methods. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Husse B and Isenberg G: Cyclic mechanical
strain causes cAMP-response element binding protein activation by
different pathways in cardiac fibroblasts. Heart Int.
5:e32010.PubMed/NCBI
|
|
11
|
Yoshida K and Matsuzaki K: Differential
regulation of TGF-β/Smad signaling in hepatic stellate cells
between acute and chronic liver injuries. Front Physiol. 3:532012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ran I, Laplante I and Lacaille JC:
CREB-dependent transcriptional control and quantal changes in
persistent long-term potentiation in hippocampal interneurons. J
Neurosci. 32:6335–6350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Michael LF, Asahara H, Shulman AI, Kraus
WL and Montminy M: The phosphorylation status of a cyclic
AMP-responsive activator is modulated via a chromatin-dependent
mechanism. Mol Cell Biol. 20:1596–1603. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wen AY, Sakamoto KM and Miller LS: The
role of the transcription factor CREB in immune function. J
Immunol. 185:6413–6419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xing D and Bonanno JA: Effect of cAMP on
TGFbeta1-induced corneal keratocyte-myofibroblast transformation.
Invest Ophthalmol Vis Sci. 50:626–633. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ding N, Yu RT, Subramaniam N, Sherman MH,
Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, et al: A
vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic
response. Cell. 153:601–613. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chung AC, Dong Y, Yang W, Zhong X, Li R
and Lan HY: Smad7 suppresses renal fibrosis via altering expression
of TGF-β/Smad3-regulated microRNAs. Mol Ther. 21:388–398. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Fan D, Wang C, Wang JY, Cui XB, Wu
D, Zhou Y and Wu LL: Angiotensin II increases periostin expression
via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac
fibroblasts. Cardiovasc Res. 91:80–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cho IJ, Kim YW, Han CY, Kim EH, Anderson
RA, Lee YS, Lee CH, Hwang SJ and Kim SG: E-cadherin antagonizes
transforming growth factor β1 gene induction in hepatic stellate
cells by inhibiting RhoA-dependent Smad3 phosphorylation.
Hepatology. 52:2053–2064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee KS, Park SJ, Kim SR, Min KH, Lee KY,
Choe YH, Hong SH, Lee YR, Kim JS, Hong SJ and Lee YC: Inhibition of
VEGF blocks TGF-beta1 production through a PI3K/Akt signalling
pathway. Eur Respir J. 31:523–531. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Li Y, Chen M, Su X, Yi D, Lu P
and Zhu D: 15-LO/15-HETE mediated vascular adventitia fibrosis via
p38 MAPK-dependent TGF-β. J Cell Physiol. 229:245–257. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ding ZY, Jin GN, Liang HF, Wang W, Chen
WX, Datta PK, Zhang MZ, Zhang B and Chen XP: Transforming growth
factor β induces expression of connective tissue growth factor in
hepatic progenitor cells through Smad independent signaling. Cell
Signal. 25:1981–1992. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang L, Inokuchi S, Roh YS, Song J, Loomba
R, Park EJ and Seki E: Transforming growth factor-β signaling in
hepatocytes promotes hepatic fibrosis and carcinogenesis in mice
with hepatocyte-specific deletion of TAK1. Gastroenterology.
144:1042–1054.e4. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kasper LH, Lerach S, Wang J, Wu S, Jeevan
T and Brindle PK: CBP/p300 double null cells reveal effect of
coactivator level and diversity on CREB transactivation. EMBO J.
29:3660–3672. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang J, Weaver IC, Gauthier-Fisher A, Wang
H, He L, Yeomans J, Wondisford F, Kaplan DR and Miller FD: CBP
histone acetyltransferase activity regulates embryonic neural
differentiation in the normal and Rubinstein-Taybi syndrome brain.
Dev Cell. 18:114–125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen W, Lam SS, Srinath H, Schiffer CA,
Royer WE Jr and Lin K: Competition between Ski and CREB-binding
protein for binding to Smad proteins in transforming growth
factor-beta signaling. J Biol Chem. 282:11365–11376. 2007.
View Article : Google Scholar : PubMed/NCBI
|