Introduction
Cigarette smoking is a major cause of premature
mortality rates worldwide, and is an established risk factor for
cardiovascular disease (1).
Tobacco smoke contains >4,000 chemical components (2), which have been shown to exert marked
effects on the magnitude and regulation of inflammatory responses
(3–5). Compounds, including nicotine, act via
various pathways to cause significant damage to endothelial cells
(6,7), resulting in endothelial dysfunction
and a series of subsequent pathophysiological reactions, ultimately
leading to atherosclerotic vascular disease. However, the mechanism
linking nicotine to this vascular disease remains to be fully
elucidated.
Perivascular adipose tissue (PVAT) is a major
endocrine and paracrine organ producing a variety of mediator
proteins, signaling proteins, known collectively as ‘adipokines’.
Adipocytes are the predominant cell type in adipose tissue, which
secrete numerous adipokines into the blood. It has become clear
that several adipokines are mediators, which form communications
between adipose tissues and the vasculature (8,9).
A number of adipokines are mediators, which allow
for communication between adipose tissues and the vasculature. PVAT
can secrete inflammatory adipokines, including tumor necrosis
factor (TNF)-α, interleukin (IL)-6 and IL-1β, however, certain
adipokines can also secrete anti-inflammatory adipokines, including
adiponectin and IL-10. Adiponectin is a polypeptide and an adipose
tissue-specific collagen-like factor, which is abundantly present
in plasma and possesses anti-atherogenic properties. Adiponectin
has been associated with cardiovascular disease (10) and hypertension (11). There is also evidence indicating
that adipokines can alter endothelial function and atherogenesis. A
study of the association between adiponectin concentrations and
smoking habits indicated that the nicotine in tobacco smoke can
reduce plasma adiponectin via inhibition of the secretion and
expression of adiponectin in adipocytes (12). When these inflammatory adipokines
are released from adipose tissue, they induce adverse effects on
the vasculature (13) and induce
the release of adhesion molecules by endothelial cells, thereby
promoting leukocyte-endothelium interactions during the
inflammatory response. These observations indicate that PVAT
malfunction can further exacerbate endothelial inflammation via
abnormal cytokine secretion. However, whether nicotine can cause
abnormal inflammatory responses in the vessel wall through altering
the endocrine and paracrine functions of PVAT, which can eventually
result in increased endothelial inflammation, remains to be fully
elucidated.
The present study investigated the roles of
nicotine-induced secretion of inflammatory cytokines and
adiponectin from adipocytes and endothelial cells, and examined the
effects on interactions between adipocytes and endothelial cells.
The nicotine-induced stimulated secretion of adiponectin and
inflammatory adipokines by adipocytes and endothelial cells were
examined. A co-culture system of adipocytes and endothelial cells
was also included to examine the effects of nicotine-induced
secretion of inflammatory cytokines and adiponectin on the
expression of adhesion molecules. It was shown that nicotine
enhanced the secretion of inflammatory cytokines and the expression
of adhesion molecules in co-cultures of adipocytes and endothelial
cells.
Materials and methods
Materials
Dulbecco's modified Eagle's medium (DMEM) containing
high glucose and glutamine was purchased from Hyclone (GE
Healthcare Life Sciences, Logan, UT, USA), and fetal bovine serum
(FBS), calf serum (CS), trypsin and penicillin G-streptomycin were
purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham, MA,
USA). Insulin, 3-isobutyl-1-methylxanthine, and dexamethasone were
obtained from Sigma-Aldrich (Merck Millipore, Darmstadt, Germany).
Endothelial cell medium (ECM) and endothelial cell growth
supplement (ECGS) were purchased from ScienCell Research
Laboratories (Carlsbad, CA, USA). The Transwell system was
purchased from Corning Incorporated (Corning, NY, USA). Anti-VCAM-1
antibody was purchased from Epitomics (Burlingame, CA, USA).
Anti-ICAM-1 and anti-NF-κB p65 were from Abcam (Cambridge, UK). The
horseradish peroxidase (HRP)-conjugated secondary antibodies were
from Abcam, and the enhanced chemiluminescence assay kit was from
Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). ELISA kits
for IL-1β, IL-6, TNF-α and adiponectin were obtained from R&D
Systems, Inc. (Minneapolis, MN, USA). Nicotine and all other
chemicals were purchased from Sigma-Aldrich (Merck Millipore).
Cell culture and treatment
Mouse 3T3-L1 preadipocytes (Cell Resource Center,
Institute of Basic Medical Sciences, Chinese Academy of Medical
Sciences and Peking Union Medical College, Beijing, China) were
cultured in DMEM-high glucose basal medium with 10% CS and
antibiotics (100 U/ml penicillin G and 100 µg/ml streptomycin) in a
humidified atmosphere containing 5% CO2 at 37°C. The
culture medium was replaced every 48 h. To obtain fully
differentiated adipocytes, on day 2 post-confluence, the cells were
induced to differentiate using a standard protocol, as described
previously (14). In brief, the
cells were induced to differentiate by replacement of the medium to
DMEM containing 10% FBS supplemented with an adipogenic cocktail (1
µg/ml insulin, 0.5 mM isobutylmethylxanthine and 1 mM
dexamethasone). After 2 days, the medium was replaced with
DMEM-high glucose medium supplemented with 10% FBS and insulin. The
culture medium was then replaced every 2 days with DMEM-high
glucose with 10% FBS medium until >90% of cells were observed to
exhibit an adipocyte phenotype when observed under a light
microscope (Olympus Corporation, Tokyo, Japan).
Human umbilical vein endothelial cells (HUVECs;
ScienCell Research Laboratories) were maintained in ECM and 15 mg/l
ECGS in an atmosphere of 5% CO2 at 37°C.
The standard protocol used for the mouse 3T3-L1
preadipocytes and HUVECs was similar to that described previously
(15,16), and the mouse 3T3-L1 preadipocyte
differentiation protocol was as described above. The mature
adipocytes and HUVECs were co-cultured in a 6-well Transwell system
with a 0.4-µm porous membrane to separate the upper and lower
chambers. An initial seeding density of 1×105
differentiated 3T3-L1 cells per well were cultured in the lower
chamber and the HUVECs density of 5×104 were cultured in
the upper chamber.
The mature adipocytes and HUVECs were exposed to
nicotine concentrations of 10−6, 10−7 and
10−8 mol/l for 24 h, following which the supernatant and
cells were collected. The culture supernatants were collected from
each sample and centrifuged at 800 × g for 5 min at 4°C.
Western blot analysis
Total proteins were extracted from the mature
adipocytes and HUVECs following cell lysis, the lysates were
maintained at 100°C for 10 min and centrifuged at 23,200 × g
for 15 min. The protein concentrations in each sample were
determined using a Bicinchoninic Acid Protein Assay kit (Beyotime
Institute of Biotechnology, Haimen, China) and equal quantities (20
µg) were resolved using 10% SDS-polyacrylamide gels. The proteins
were transferred onto a polyvinylidene fluoride membrane (EMD
Millipore, Billerica, MA, USA). The membranes were blocked in 5%
skimmed milk in TBST containing 20 mM, Tris-HCl (pH 7.6), 137 mM
NaCl and 0.05% Tween-20, for 1 h at room temperature. The membranes
were then incubated with rabbit polyclonal antibody against VCAM-1
(1:1,000; cat. no. ab134047), rabbit polyclonal antibody against
ICAM-1 (1:1,000; cat. no. ab7815), or rabbit polyclonal antibody
against NF-κB p65 (1:1,000; cat. no. ab16502) overnight at 4°C.
Subsequently, the membranes were incubated with HRP-conjugated
secondary antibodies (1:5,000; cat. no. ab205718) for 1 h at room
temperature. Bands were detected using an enhanced
chemiluminescence assay kit.
ELISA
The culture supernatants from the mature adipocytes,
HUVECs and the co-cultured cells in the Transwell system were
collected from each sample 24 h following the addition of nicotine,
and supernatants were then centrifuged at 800 × g for 5 min
at 4°C. The collected supernatants were aliquoted, snap frozen and
stored at −80°C until later use. Concentrations of IL-1β, IL-6,
TNF-α, and adiponectin were assayed using mouse ELISA kits for
IL-1β, IL-6, TNF-α and adiponectin according to the manufacturer's
protocols.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean and were compared using analysis of variance. Each
experiment was performed at least three times. P<0.05 was
considered to indicate a statistically significant difference.
Statistical analysis was performed using SPSS 17.0 software (SPSS,
Inc., Chicago, IL, USA).
Results
Effects of nicotine on the secretion
of adiponectin and inflammatory adipokines in 3T3-L1
adipocytes
The present study first examined the effects of
nicotine on the secretion of adipokines by 3T3-L1 adipocytes.
Mature adipocytes were exposed to various concentrations of
nicotine (final concentrations, 10−6, 10−7
and 10−8 mol/l) for 24 h.
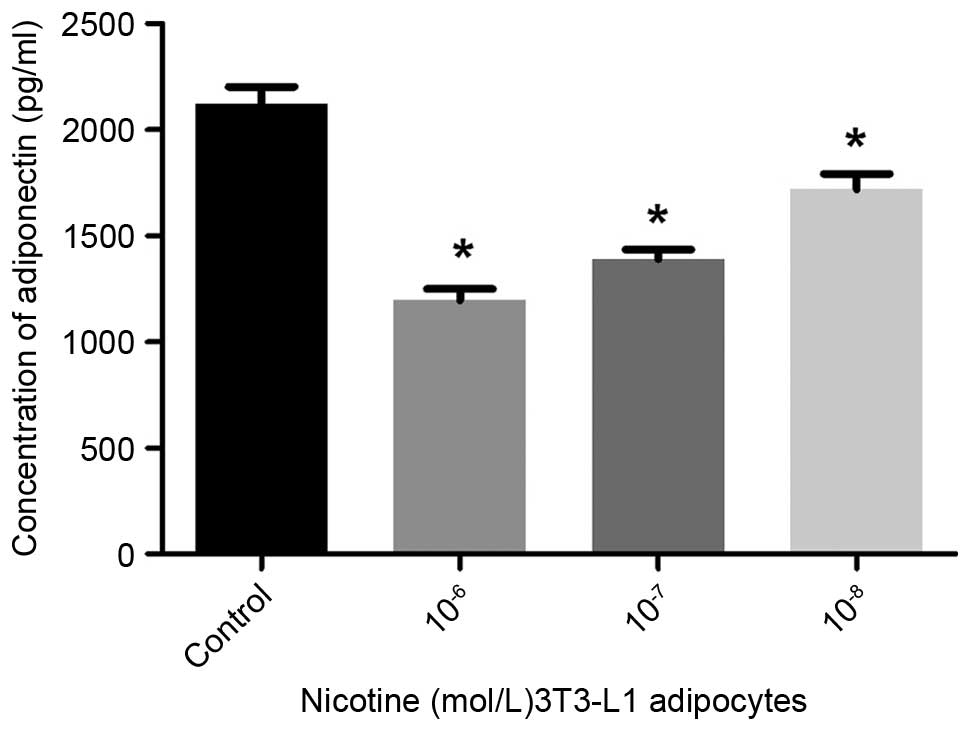
Compared with the controls, the secretion of
adiponectin into the culture media was significantly reduced
following treatment with nicotine at concentrations of
10−6, 10−7 and 10−8 mol/l
(P<0.05; Fig. 1). The secretion
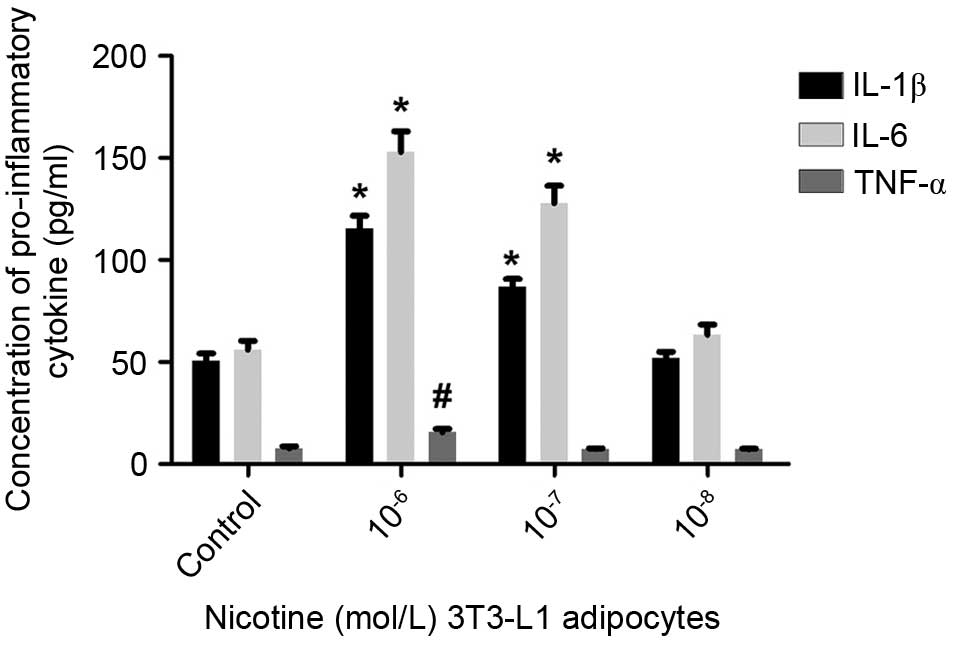
levels of IL-1β and IL-6 were then examined using ELISA, and it was
found that 10−6 or 10−7 mol/l nicotine
resulted in elevated levels of IL-1β and IL-6 (Fig. 2). Following treatment with
10−7 or 10−8 mol/l nicotine, no significant
difference in the secretion of TNF-α by mature adipocytes was
observed (P>0.05); only at concentrations of 10−6
mol/l nicotine did the levels of secreted TNF-α increase
significantly (P<0.05). These findings suggested that nicotine
induced a reduction in the levels of adiponectin, and increases in
the levels of IL-1β and IL-6 secreted by mature adipocytes.
Effects of nicotine on inflammatory
cytokine secretion by HUVECs
The supernatants from HUVECs were collected at a
single time point (24 h) and exposed to various concentrations of
nicotine (final concentrations, 10−6, 10−7
and 10−8 mol/l).
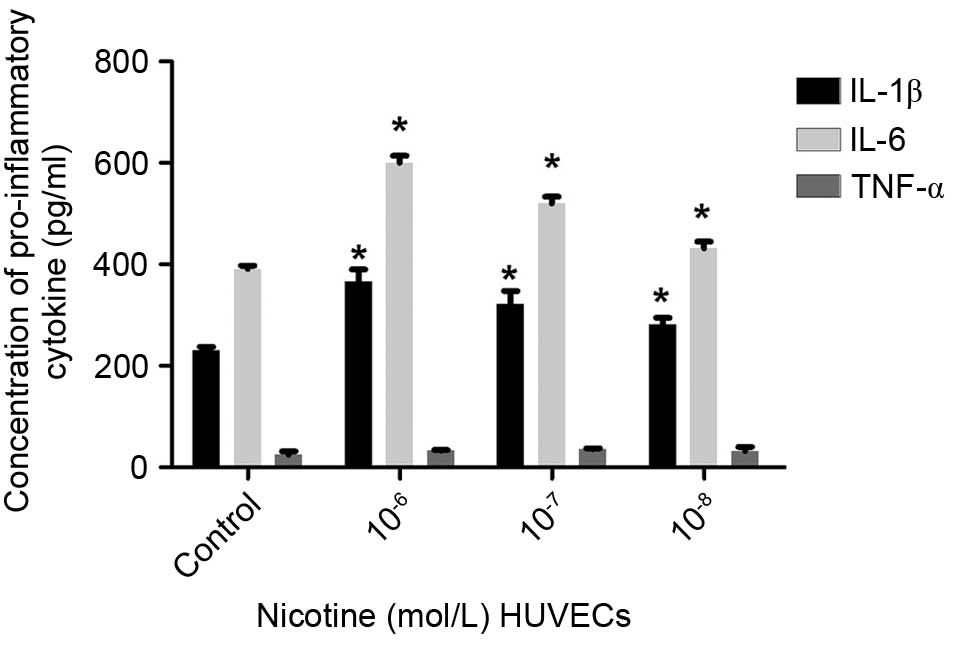
The levels of IL-1β, IL-6 and TNF-α were detected
using ELISA. The secretion of IL-1β and IL-6 by HUVECs was elevated
following nicotine treatment, compared with the control cells
(P<0.05), whereas no significant differences were observed in
the secretion of TNF-α (Fig. 3).
These findings indicated that the secretion of IL-1β and IL-6 by
HUVECs showed a similar trend to that of the mature adipocytes,
suggesting a causative association between nicotine and the
secretion of inflammatory adipokines by adipocytes and HUVECs.
Nicotine-induced expression of
inflammatory adipokines, adhesion molecules and adiponectin, and
their effects on adipocyte-HUVEC interactions
The co-cultured mature adipocytes and HUVECs were
exposed to 10−6 mol/l nicotine for 24 h. This co-culture
of the mature adipocytes and HUVECs resulted in a significant
increase in the levels of IL-6 and IL-1β in the supernatants
(P<0.05; Fig. 4). The levels of
TNF-α were similar to those of the single cultures of mature
adipocytes and HUVECs, but a statistically significant difference
was observed (P<0.05; Fig. 4).
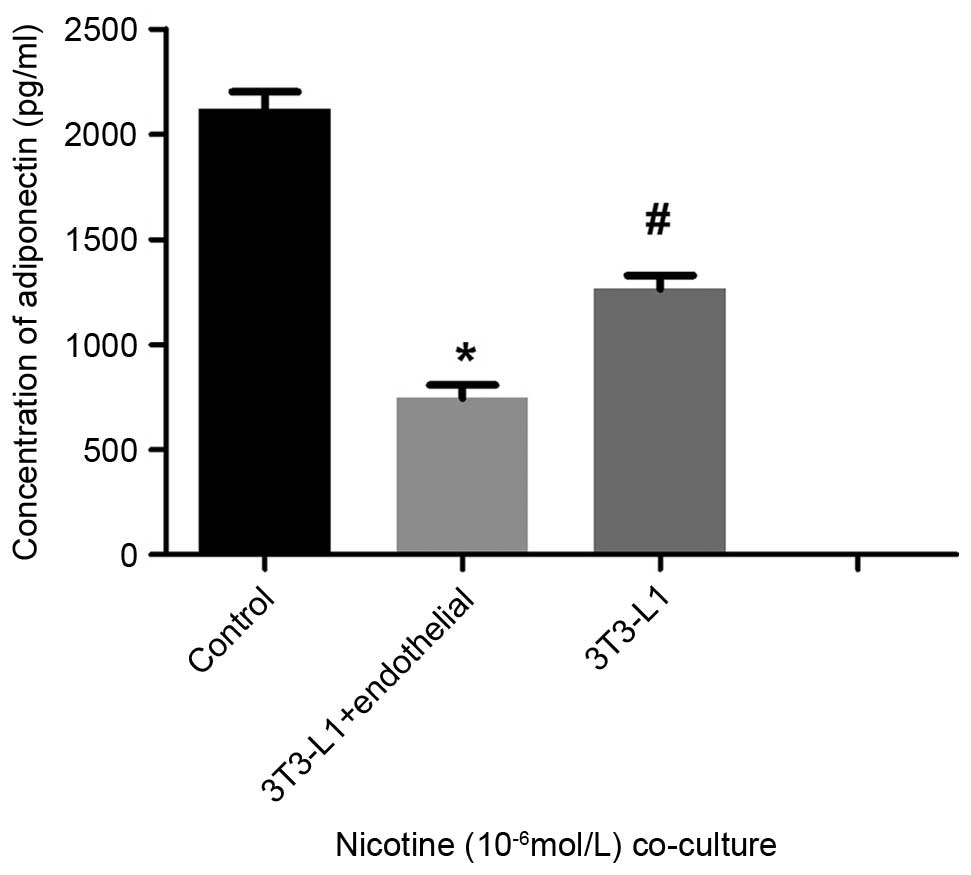
Furthermore, measurements of secreted adiponectin using ELISA
showed that the levels in the co-culture group were significantly
decreased, compared with those of the mature adipocytes cultured
alone (P<0.05; Fig. 5). Western
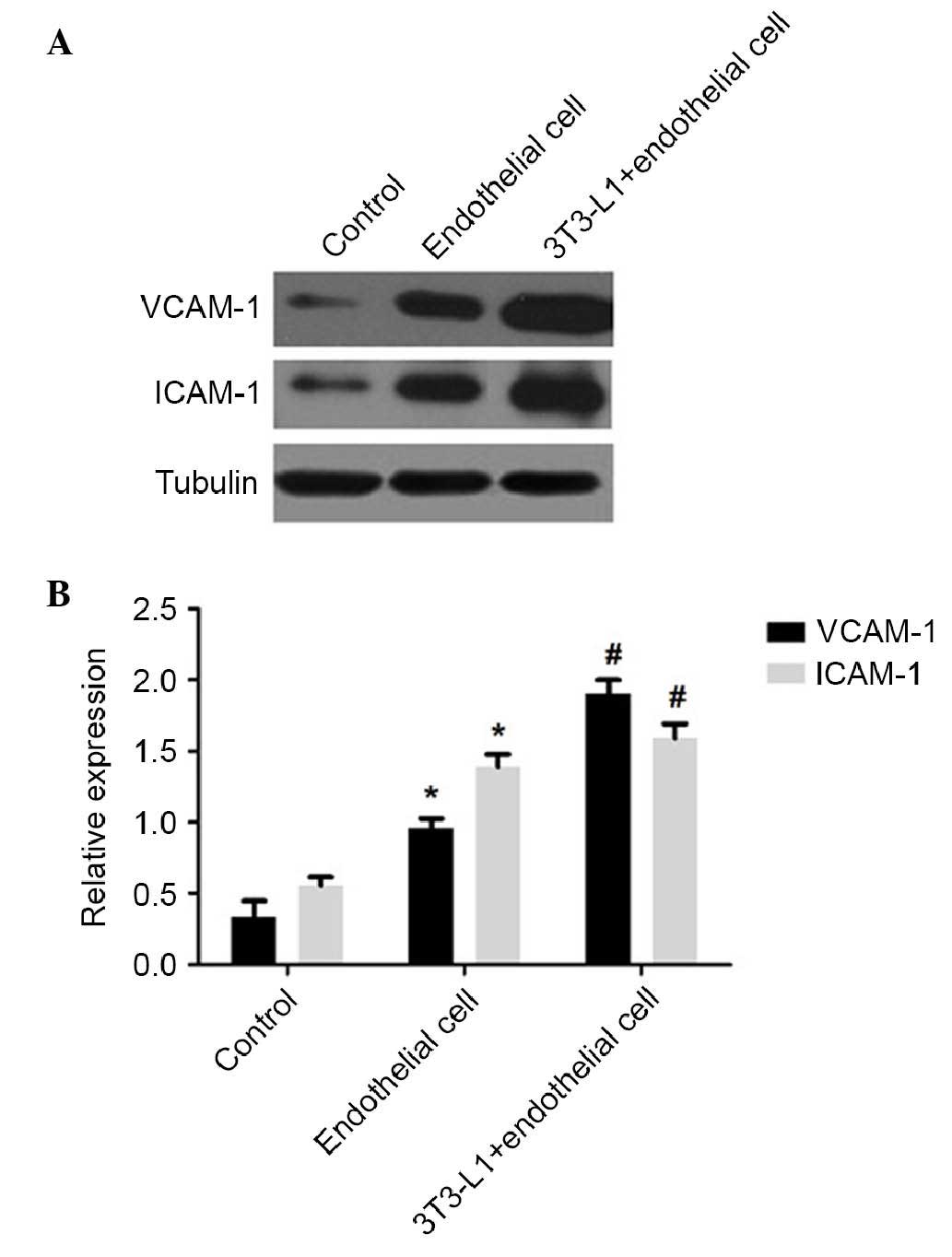
blot analysis showed that nicotine induced the expression of
VCAM-1, ICAM-1 and NF-κB p65 in the co-cultures of mature
adipocytes and HUVECs. The levels of VCAM-1 and ICAM-1 in the
co-cultured cells were significantly elevated, compared with those
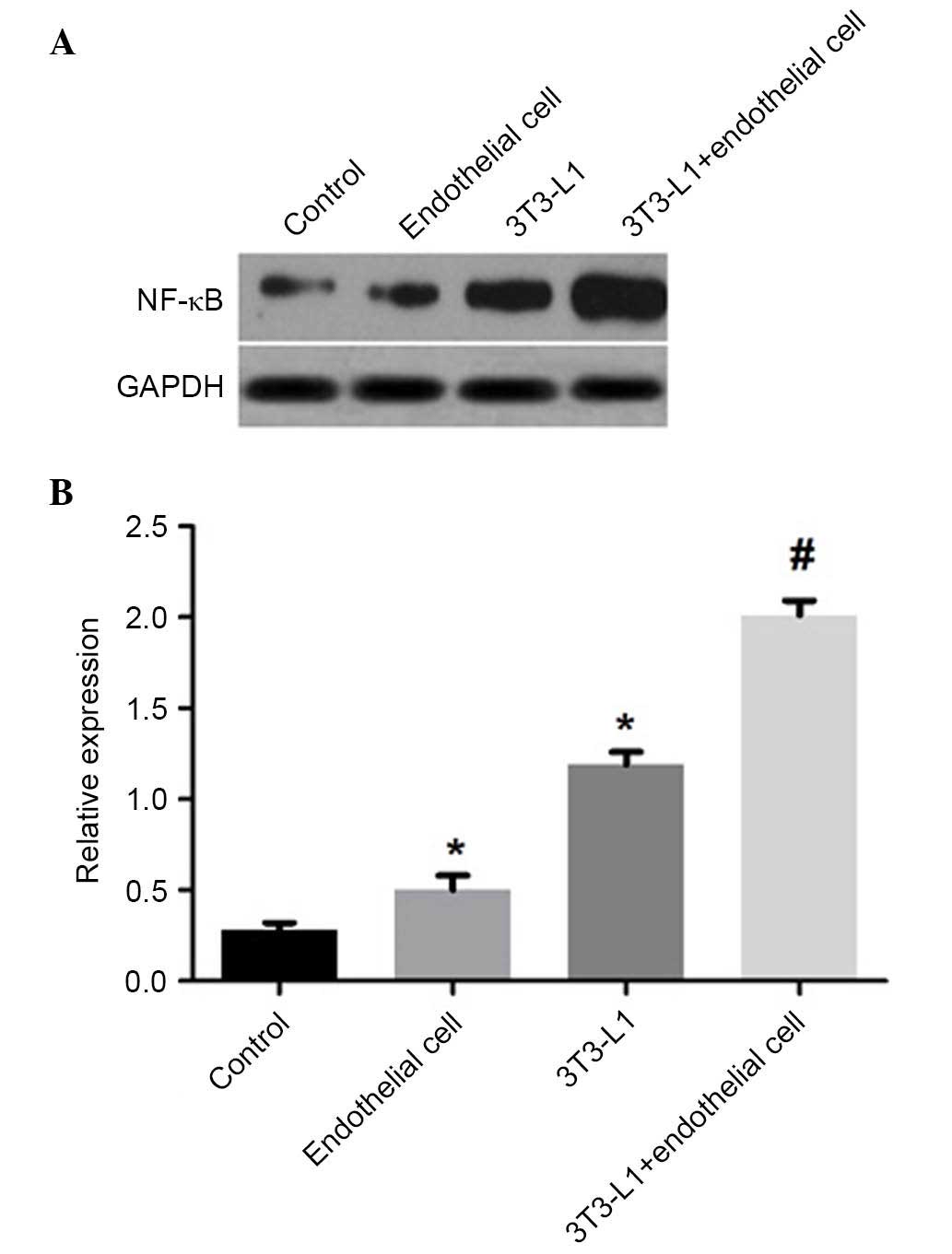
in the HUVECs cultured alone (P<0.01; Fig. 6), and the co-culture group had
significantly higher levels of NF-κB p65, compared with the mature
adipocytes or HUVECs cultured alone (P<0.01; Fig. 7). These findings suggested that
nicotine markedly upregulated the expression of inflammatory
adipokines and adhesion molecules in co-cultured mature adipocytes
and HUVECs. Additionally, the levels of adiponectin were
significantly reduced in the co-cultures, compared with the levels
in the mature adipocytes cultured alone.
Discussion
Epidemiological studies have shown that smoking is a
major cause of atherosclerosis. Smoking can cause endothelial
dysfunction through various mechanisms. PVAT can affect the
structure and function of blood vessel walls by altering their
endocrine and paracrine functions. The present study investigated
nicotine stimulation of mature adipocytes, endothelial cells and a
co-culture of these two cell types to investigate how adipocytes
affect endocrine and paracrine functions to accelerate endothelial
inflammation.
It has been shown that smoking habits are associated
with adiponectin concentrations in men, in cultured 3T3-L1
adipocytes, nicotine reduces the secretion and expression of
adiponectin (17). Gao et
al (18) reported that
nicotine can alter the composition and function of PVAT, resulting
in increased blood pressure. Therefore, the present study
investigated whether nicotine can cause vessel wall abnormalities
as a consequence of the inflammatory response by altering the
endocrine and paracrine functions of PVAT. Mature adipocytes were
exposed to various concentrations of nicotine (10−6,
10−7 and 10−8 mol/l) for 24 h. It was found
that the expression levels of IL-6 and IL-1β were significantly
elevated in the nicotine-treated group. No significant differences
were found in the levels of TNF-α following stimulation with
10−7 or 10−8 mol/l nicotine, and only at a
concentration of 10−6 mol/l nicotine did the secretion
of TNF-α increase significantly. Concentrations of plasma nicotine
have been reported to approach 10−7 mol/l in smokers
(19). A previous study showed
that HUVECs secrete TNF-α in response to nicotine at levels similar
to those found in the serum following smoking
(10−6-10−10 mol/l), although the activity of
TNF-α returned to baseline levels by 24 h (20). Following stimulation of adipocytes
with nicotine for 24 h in the present study, the expression levels
of TNF-α showed a similar trend to those of the HUVECs. Adiponectin
is an anti-inflammatory cytokine, which can be secreted by
adipocytes. It can inhibit inflammation by reducing the expression
of various adhesion molecules by endothelial cells (21,22).
The findings of the present study showed that nicotine reduced the
secreted levels of adiponectin in mature adipocytes, similar to
findings reported in a previous study (19). Together, the findings of the
present study suggested that nicotine at concentrations similar to
those observed in the serum of smokers can lead to PVAT
dysfunction, and cause the abnormal secretion of adiponectin and
inflammatory adipokines. This suggested that PVAT may contribute to
nicotine-induced endothelial inflammatory responses.
In this present study, it was also shown that
secretion of the IL-6 and IL-1β inflammatory adipokines by HUVECs
was significantly increased following 24 h treatment with nicotine
at concentrations of 10−6-10−8 mol/l,
however, no significant difference was found in the levels of
TNF-α. Previous studies have shown that nicotine can stimulate the
secretion of TNF-α and IL-1β by macrophages, and promote the
expression of endothelial cell adhesion factor (23). Overall, the results of the present
study suggested that nicotine stimulation of endothelial cells
caused increased secretion of inflammatory adipokines and increased
endothelial inflammation.
The results described above showed that nicotine
caused the abnormal secretion of adiponectin by adipocytes and that
endothelial cells secreted inflammatory adipokines. To investigate
the interactions between these cell types, adipocyte and HUVEC
co-cultures were examined, which were exposed to 10−6
mol/l nicotine for 24 h. The co-culture led to upregulation in the
secretion of IL-6, IL-1β and TNF-α, however, the levels of
adiponectin were decreased in the co-culture supernatants.
Additionally, it was revealed that, in the co-culture group, the
secretion of TNF-α was enhanced by stimulation with
adipocyte-conditioned media. As the co-culture resulted in levels
of adiponectin similar to those of the adipocytes cultured alone,
further reductions in the secretion of adiponectin by adipocytes
appeared to affect interactions with endothelial cells and the
secretion of inflammatory adipokines. VCAM-1 and ICAM-1 can
contribute to the recruitment of circulating inflammatory cells
into vascular walls, which may explain the nicotine-induced
infiltration of inflammatory cells. The findings of the present
study suggested that the expression levels of ICAM-1 and VCAM-1
were upregulated in the co-culture system. A previous study also
reported nicotine-induced expression of ICAM-1 and VCAM-1 by
endothelial cells (24). Several
factors may be involved in promoting inflammatory responses in
endothelial cells, among which NF-κB is an important mediator of
tissue inflammation. This transcription factor can drive several
aspects of inflammation and induce the expression of several
inflammatory mediators, including ICAM-1, VCAM-1 and various
interleukins. A number of studies have indicated that cigarette
smoking can cause tissue inflammation via the induction of NF-κB,
and nicotine has been reported to upregulate the expression of
NF-κB in rat intravascular tissues (25–27).
In the present study, nicotine markedly upregulated the expression
of NF-κB in the co-cultured cells. In previously reported in
vivo experiments, adiponectin has been found to modulate the
inflammatory response of endothelial cells via cross-talk between
the cyclic-AMP-protein kinase A and NF-κB signaling pathways
(28). Therefore, the present
study hypothesized that nicotine stimulated the co-cultured cells
to activate the NF-κB signaling pathway by upregulating the
expression of NF-κB, which aggravated endothelial cells and
exacerbated an inflammatory response. The results of the present
study supported those of previous reports that adiponectin is an
important mediator in the cross-talk between adipose tissue and the
vasculature (29), therefore,
adipocyte-endothelial cell interactions may be important in
nicotine-induced endothelial inflammation.
In conclusion, the present study found that nicotine
induced adipocyte dysfunction and caused the abnormal secretion of
adiponectin and inflammatory adipokines, which further exacerbated
endothelial inflammation. These findings provide evidence for an
association between nicotine, PAVT and vascular damage, and offer
insight into PVAT-targeted therapy for the protection of
vasculature.
Acknowledgements
The present study was supported by the Peking Union
Medical College Hospital Medical Research Funds (grant no. PUMCH
001).
References
|
1
|
Ambrose JA and Barua RS: The
pathophysiology of cigarette smoking and cardiovascular disease: An
update. J Am Coll Cardiol. 43:1731–1737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Borgerding M and Klus H: Analysis of
complex mixtures-cigarette smoke. Exp Toxicol Pathol 57 (Suppl 1).
43–73. 2005. View Article : Google Scholar
|
|
3
|
Lee J, Taneja V and Vassallo R: Cigarette
smoking and inflammation: Cellular and molecular mechanisms. J Dent
Res. 91:142–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Orosz Z, Csiszar A, Labinskyy N, Smith K,
Kaminski PM, Ferdinandy P, Wolin MS, Rivera A and Ungvari Z:
Cigarette smoke-induced proinflammatory alterations in the
endothelial phenotype: Role of NAD (P)H oxidase activation. Am J
Physiol Heart Circ Physiol. 292:H130–H139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arnson Y, Shoenfeld Y and Amital H:
Effects of tobacco smoke on immunity, inflammation and
autoimmunity. J Autoimmun. 34:J258–J265. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kuhlmann CR, Trümper JR, Tillmanns H,
Schaefer C Alexander and Erdogan A: Nicotine inhibits large
conductance Ca(2+)-activated K(+) channels and the NO/−cGMP
signaling pathway in cultured human endothelial cells. Scand
Cardiovasc J. 39:348–352. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Glantz SA and Parmley WW: Passive and
active smoking. A problem for adults. Circulation. 94:596–598.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Halberg N, Wernstedt-Asterholm I and
Scherer PE: The adipocyte as an endocrine cell. Endocrinol Metab
Clin North Am. 37:753–768, x-xi. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lehr S, Hartwig S and Sell H: Adipokines:
A treasure trove for the discovery of biomarkers for metabolic
disorders. Proteomics Clin Appl. 6:91–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kumada M, Kihara S, Sumitsuji S, Kawamoto
T, Matsumoto S, Ouchi N, Arita Y, Okamoto Y, Shimomura I, Hiraoka
H, et al: Association of hypoadiponectinemia with coronary artery
disease in men. Arterioscler Thromb Vasc Biol. 23:85–89. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Adamczak M, Wiecek A, Funahashi T, Chudek
J, Kokot F and Matsuzawa Y: Decreased plasma adiponectin
concentration in patients with essential hypertension. Am J
Hypertens. 16:72–75. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ouchi N, Ohishi M, Kihara S, Funahashi T,
Nakamura T, Nagaretani H, Kumada M, Ohashi K, Okamoto Y, Nishizawa
H, et al: Association of hypoadiponectinemia with impaired
vasoreactivity. Hypertension. 42:231–234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Van de Voorde J, Pauwels B, Boydens C and
Decaluwé K: Adipocytokines in relation to cardiovascular disease.
Metabolism. 62:1513–1521. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suganami T, Tanimoto-Koyama K, Nishida J,
Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S,
et al: Role of the toll-like receptor 4/NF-kappaB pathway in
saturated fatty acid-induced inflammatory changes in the
interaction between adipocytes and macrophages. Arterioscler Thromb
Vasc Biol. 27:84–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamashita A, Soga Y, Iwamoto Y, Yoshizawa
S, Iwata H, Kokeguchi S, Takashiba S and Nishimura F:
Macrophage-adipocyte interaction: Marked interleukin-6 production
by lipopolysaccharide. Obesity (Silver Spring). 15:2549–2552. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lai N, Jayaraman A and Lee K: Enhanced
proliferation of human umbilical vein endothelial cells and
differentiation of 3T3-L1 adipocytes in coculture. Tissue Eng Part
A. 15:1053–1061. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iwashima Y, Katsuya T, Ishikawa K, Kida I,
Ohishi M, Horio T, Ouchi N, Ohashi K, Kihara S, Funahashi T, et al:
Association of hypoadiponectinemia with smoking habit in men.
Hypertension. 45:1094–1100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao YJ, Holloway AC, Su LY, Takemori K, Lu
C and Lee RM: Effects of fetal and neonatal exposure to nicotine on
blood pressure and perivascular adipose tissue function in adult
life. Eur J Pharmacol. 590:264–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Thielen A, Klus H and Müller L: Tobacco
smoke: Unraveling a controversial subject. Exp Toxicol Pathol.
60:141–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Albaugh G, Kann B, Strande L, Vemulapalli
P, Hewitt C and Alexander JB: Nicotine induces endothelial
TNF-alpha expression, which mediates growth retardation in vitro. J
Surg Res. 99:381–384. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Deng G, Long Y, Yu YR and Li MR:
Adiponectin directly improves endothelial dysfunction in obese rats
through the AMPK-eNOS pathway. Int J Obes (Lond). 34:165–171. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Y, Wang X, Lau WB, Yuan Y, Booth D,
Li JJ, Scalia R, Preston K, Gao E, Koch W and Ma XL: Adiponectin
inhibits tumor necrosis factor-alpha-induced vascular inflammatory
response via caveolin-mediated ceramidase recruitment and
activation. Circ Res. 114:792–805. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Wang L, Ai X, Zhao J, Hao X, Lu Y
and Qiao Z: Nicotine could augment adhesion molecule expression in
human endothelial cells through macrophages secreting TNF-alpha,
IL-1beta. Int Immunopharmacol. 4:1675–1686. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ueno H, Pradhan S, Schlessel D, Hirasawa H
and Sumpio BE: Nicotine enhances human vascular endothelial cell
expression of ICAM-1 and VCAM-1 via protein kinase C, p38
mitogen-activated protein kinase, NF-kappaB, and AP-1. Cardiovasc
Toxicol. 6:39–50. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gonçalves RB, Coletta RD, Silvério KG,
Benevides L, Casati MZ, da Silva JS and Nociti FH Jr: Impact of
smoking on inflammation: Overview of molecular mechanisms. Inflamm
Res. 60:409–424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tsiara S, Elisaf M and Mikhailidis DP:
Influence of smoking on predictors of vascular disease. Angiology.
54:507–530. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yanbaeva DG, Dentener MA, Creutzberg EC,
Wesseling G and Wouters EF: Systemic effects of smoking. Chest.
131:1557–1566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ouchi N, Kihara S, Arita Y, Okamoto Y,
Maeda K, Kuriyama H, Hotta K, Nishida M, Takahashi M, Muraguchi M,
et al: Adiponectin, an adipocyte-derived plasma protein, inhibits
endothelial NF-kappaB signaling through a cAMP-dependent pathway.
Circulation. 102:1296–1301. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li FY, Cheng KK, Lam KS, Vanhoutte PM and
Xu A: Cross-talk between adipose tissue and vasculature: Role of
adiponectin. Acta Physiol (Oxf). 203:167–180. 2011. View Article : Google Scholar : PubMed/NCBI
|