Introduction
Alzheimer's disease (AD) is the most frequent cause
of dementia in the elderly. The global incidence rates of dementia
reached ~24 million in 2012, and is predicted to double every 20
years until the year 2040 (1).
Therefore, the prevention and treatment of AD will become a
significant economic burden on public health services in the
future. As a progressive neurodegenerative disorder, AD is
characterized by the accumulation of β-amyloid (Aβ) peptides,
hyperphosphorylated tau proteins and neuronal loss. Resistance of
amyloid deposition and tau protein aggregation has been the primary
focus of research into the treatment of AD in recent decades.
Although the exact etiology of AD remains unclear, neuronal
apoptosis is a primary reason for the loss of neurons (2). Amyloid cascade theory is currently
the leading theory for the etiology of AD (3). Immunotherapeutic methods have been
demonstrated to reduce amyloid-induced cytotoxicity and
neurodegeneration (4). The most
notable of these immunotherapies are bapineuzumab and solanezumab.
However, the results of phase III trials involving anti-amyloid
treatment of AD with solanezumab and bapineuzumab have yielded
disappointing results (5,6). Karran and Hardy (7) dismiss the overwhelming evidence
obtained over the past few years demonstrating that anti-Aβ therapy
to remove brain Aβ deposits in patients with AD was a clinical
failure. The results of these phase III clinical trials involving
solanezumab and bapineuzumab demonstrate that AD is a complex
disease, involving a number of pathogenic factors. The use of
anti-amyloid therapies for the treatment of AD remains
controversial. Therefore, a combination of strategies to treat AD
is required. Despite the known role of N-methyl-D-aspartate
receptor antagonists and acetylcholinesterase inhibitors in
improving cognitive function and delaying the progression of
cognitive dysfunction, further exploration of the development of
novel and more effective therapeutic strategies for AD treatment
are urgently required (8).
Aβ25–35 is a synthetic peptide constituting 11 amino
acids, which is not detected under normal physiological conditions.
However, various investigators have used it as a model for
full-length peptide studies, as it is a biologically active
fragment of Aβ and contains the structure responsible for
neurotoxicity. Previous studies have demonstrated that oxidative
stress is involved in the generation of Aβ25–35
toxicity, which in turn involves the disruption of calcium
homeostasis, oxygen radicals and nitric oxide, ultimately leading
to mitochondrial dysfunction. Mitochondrial cytochrome c release
and apoptosis-linked proteases subsequently initiate apoptosis
(9). Therefore, antioxidants may
inhibit the apoptosis induced by Aβ25–35.
κ-carrageenan-derived oligosaccharide, extracted and purified from
marine red algae, is an antioxidant (10), and exerts antiviral (11), anti-aggregant and anticoagulant
(12) effects. Compared with other
antioxidants, κ-carrageenan oligosaccharides have small molecular
weights, good solubility, and a wide range of of biological
activities. In addition, studies have demonstrated that Aβ can
directly activate the release of inflammatory cytokines in
microglia, and induce a respiratory burst to generate a large
volume of peroxide free radicals, which damage neurons (13). κ-carrageenan oligosaccharides have
been demonstrated to inhibit the excessive activation of microglia
(14). Stress-activated protein
kinases belong to the serine/threonine kinase family (15). Of these, the c-Jun N-terminal
kinases (JNKs) have been implicated in the pathogenesis of AD. The
introduction of Aβ peptides into primary cortical neuron cultures
has been demonstrated to induce JNK activation and cell death
(16). Increasing evidence
indicates that the JNK signaling pathway is activated in AD in
susceptible regions of the brain. Aβ25–35-induced
neuronal dysfunction is, in part, mediated by alterations in signal
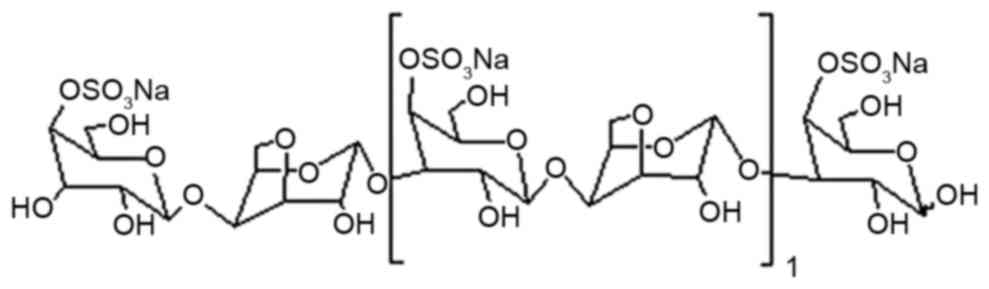
transduction pathways including JNK (17). κ-carrageenan-derived
pentasaccharide (KCP, the structure of which is presented in
Fig. 1) is a typical κ-carrageenan
oligosaccharide and, to the best of our knowledge, the
neuroprotective effect of KCP against Aβ25–35-induced
damage remains to be investigated. The present study examined the
effect of KCP on Aβ-induced apoptosis via the JNK signaling pathway
in SH-SY5Y cells.
Materials and methods
Cells and reagents
SH-SY5Y human neuroblastoma cells were donated by
the Institute of Neuroscience, Chongqing Medical University
(Chongqing, China). Aβ25–35 was purchased from Shanghai
Sangon Biotech Co., Ltd. (Shanghai, China) and was incubated at
37°C for 7 days prior to use (18). KCP was obtained from Primus Qingdao
Huili Biotechnology Co., Ltd (Qingdao, China). Low glucose
Dulbecco's modified Eagle's medium (DMEM), L-glutamine and trypsin
were purchased from HyClone (GE Healthcare Life Sciences, Logan,
UT, USA), and fetal bovine serum (FBS) was from Tianjin Hao Yang
Biological Company (Tianjin, China).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
and the Annexin V-Fluorescein Isothiocyanate (FITC)/Propidium
Iodide (PI) kit were purchased from Nanjing KenGen Biotech Co.,
Ltd. (Nanjing, China). Rabbit anti-cleaved caspase 3 (cat. no.
9664), rabbit anti-total JNK (cat. no. 9252) and rabbit
anti-phosphorylated (p)-JNK (cat. no. 9251) antibodies for western
blotting were obtained from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Mouse anti-GAPDH (cat. no. AG019), anti-mouse
horseradish peroxidase (HRP)-conjugated IgG secondary antibody
(cat. no. A0216), and anti-rabbit HRP-conjugated IgG secondary
antibody (cat. no. A0208) were obtained from Beyotime Institute of
Biotechnology (Haimen, China). Alexa Fluor®
488-conjugated goat anti-rabbit secondary antibody (cat. no.
ZF-0511) was obtained from Beijing Zhong Shan-Golden Bridge
Biological Technology Co., Ltd., (Beijing, China).
Cell culture
SH-SY5Y cells were cultured in low-glucose DMEM with
10% FBS, 1% L-glutamine, 100 U/ml penicillin and 100 µg/ml
streptomycin in a humidified atmosphere of 5% CO2 at
37°C. The media was replaced every two days. Cells that had reached
70–80% confluence were used for experiments.
Experimental protocol
To determine a suitable concentration of
Aβ25–35, cells were divided into four groups. The
control cells were cultured in normal medium, whereas the remaining
three groups were exposed to 0, 12.5, 25 or 50 µM
Aβ25–35 for 24 h. Subsequently, SH-SY5Y cells were
divided into five groups, including control, Aβ25–35
only, and three groups treated with Aβ25–35 plus 25, 50
or 100 µM KCP pre-incubated with SH-SY5Y cells for 2 h. Experiments
were performed following an incubation period of 24 h.
Cell viability assay
SH-SY5Y cells were seeded at a density of
1×104 cells/well in 96-well plates. Following overnight
drug treatment, cell viability was evaluated by the MTT assay.
Briefly, 50 µl MTT solution was added into each well, and following
a 4-h incubation at 37°C, the supernatant from each well was
carefully removed. DMSO (150 µl) was added into each well to
solubilize the formazan product. The plate was agitated for 10 min
to ensure complete dissolution of formazan. Absorbance at a
wavelength of 490 nm was measured using a SpectraMax® M2
spectrophotometer (Molecular Devices, LLC, Sunnyvale, CA, USA).
Detection of apoptosis
SH-SY5Y cells were seeded onto 6-well plates at
2×105 cells/well prior to drug treatment. The cells were
digested using trypsin without EDTA and collected at each time
point, from two wells per sample. They were subsequently washed
twice with PBS and centrifuged at 1,100 × g for 5 min at the
room temperature. The supernatant was discarded and 500 µl binding
buffer was added to the resuspended cells, followed by the addition
of 5 µl annexin V-FITC and 5 µl PI. The cells were incubated for 15
min at room temperature in the dark. The apoptotic rate was
assessed by flow cytometry using a BD Influx™ flow cytometer with
the BD FACS™ software (BD Biosciences, San Jose, CA, USA).
Western blotting
SH-SY5Y cells were seeded into T25 cell culture
flasks. Following drug treatment, cells were scraped and lysed in a
lysis buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton
X-100, 1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate,
sodium fluoride, EDTA, leupeptin and 1 mM phenylmethylsulfonyl
fluoride, and incubated on ice for 30 min. Following centrifugation
at 13,350 × g for 15 min at 4°C, the supernatant was
collected into 1.5-ml Eppendorf tubes. Protein concentrations were
determined by the bicinchoninic acid assay. Equal amounts of total
protein (40 µg) were loaded onto 10% and 12% SDS-PAGE gels, and the
resolved proteins were transferred to polyvinylidene difluoride
membranes. Membranes were blocked with 5% bovine serum albumin
(Beyotime Institute of Biotechnology) for 60 min before incubating
with primary antibodies (JNK, 1:800; p-JNK, 1:800; cleaved caspase
3, 1:1,000). GAPDH (1:800) served as a loading control. Proteins
were detected using an Enhanced Chemiluminescence reagent (Advansta
Inc., Menio Park, CA, USA). The signals were quantified using
Quantity One® software (version, 4.62; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Immunofluorescence
SH-SY5Y cells were seeded onto an 8×8 mm cell
climbing film used to cover 12-well plates. Following drug
treatment, cells were fixed with 4% paraformaldehyde at room
temperature for 20 min and washed three times with PBS. Cells were
lysed with 0.1% Triton X-100 for 10 min at 37°C, washed three times
and blocked with 4% FBS for 30 min at 37°C. Cells were subsequently
incubated with anti-cleaved caspase 3, 1:400, overnight at 4°C.
Following washing with PBS, cells were incubated with an Alexa
Fluor® 488-conjugated goat anti-rabbit secondary
antibody (dilution, 1:400) for 1 h at 37°C, washed and stained with
DAPI for 5 min. Finally, the cells were mounted with 50% glycerol
and observed under a fluorescence microscope.
Statistical analysis
Data were obtained from three separate cultures and
expressed as the mean ± standard deviation. Statistical analyses
were conducted in GraphPad Prism version 6.0 (GraphPad Software,
Inc., La Jolla, CA, USA). Data from multiple groups were compared
by one-way analysis of variance, whereas differences between two
groups were compared by Student's t-test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Effect of varying concentrations of
Aβ25–35 on cell viability
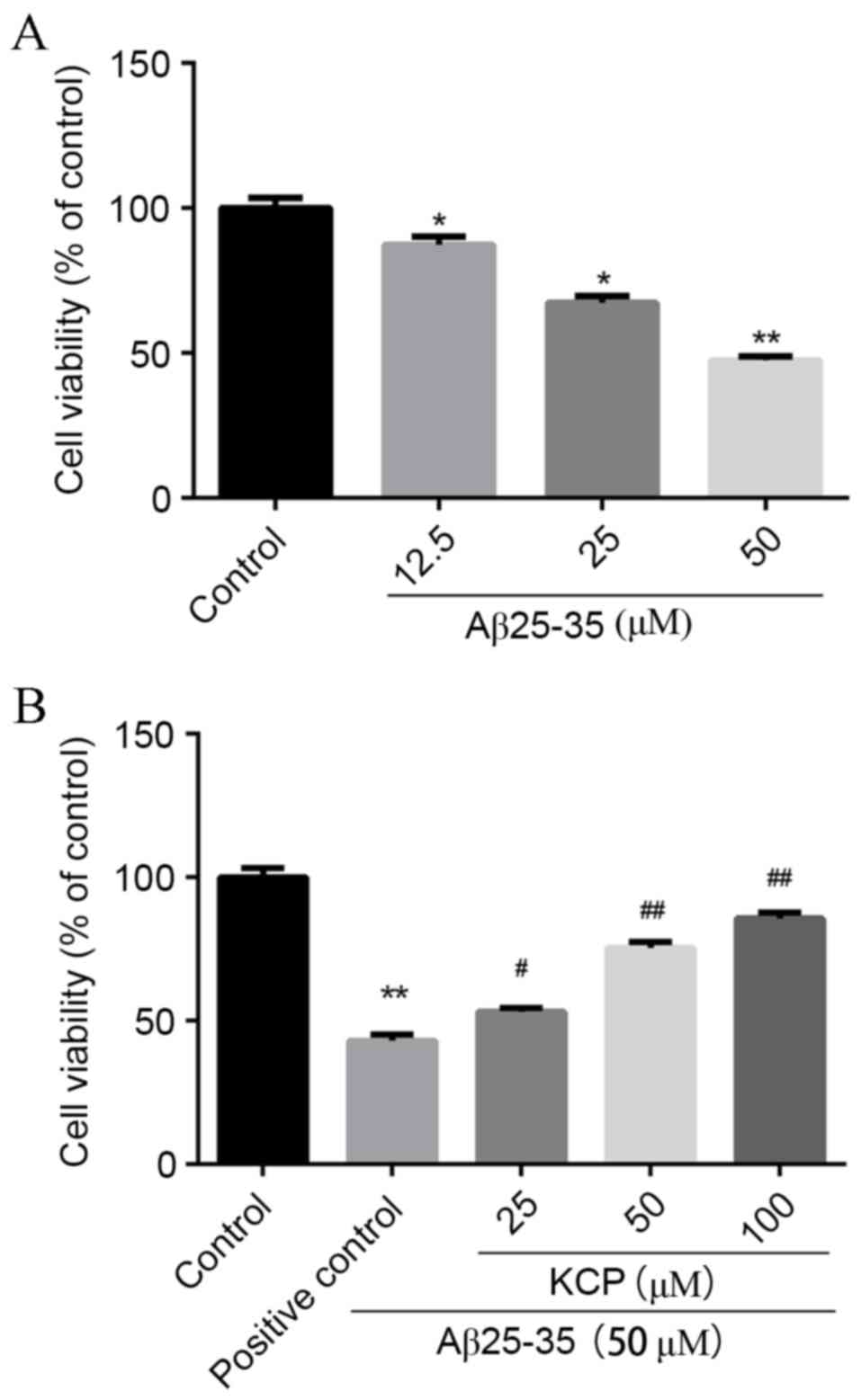
The MTT assay was used to determine
Aβ25–35-induced toxicity. SH-SY5Y cells that were
exposed to Aβ25–35 for 24 h demonstrated significant
dose-dependent cytotoxicity (Fig.
2A). Compared with untreated cells, the survival of SH-SY5Y
cells that were exposed to varying concentrations of
Aβ25–35 declined to 87.26±2.91% at 12.5 µM (P=0.0489),
67.29±2.37% at 25 µM (P=0.0015) and 47.58±1.19% at 50 µM
(P=0.0001), with a maximal difference of ~40% at a concentration of
50 µM (Fig. 2A). Therefore, 50 µM
Aβ25–35 was selected for use in subsequent
experiments.
Effect of increasing KCP
concentrations on Aβ25–35-treated cells
The MTT assay was used to determine the protective
effects of KCP against Aβ25–35-induced toxicity.
Compared with the untreated group, the survival of cells exposed to
50 µM Aβ25–35 significantly decreased to 43.08±2.08%
(P=0.001), whereas pretreatment with various concentrations of KCP
increased viability in a dose-dependent manner to 53.18±1.44%
(P=0.0162) at 25 µM, 75.38±2.23% (P=0.0004) at 50 µM and
85.70±2.12% (P=0.0001) at 100 µM (Fig.
2B). Therefore, KCP may significantly inhibit the cytotoxicity
of Aβ25–35 to improve the survival of SHSY5Y cells.
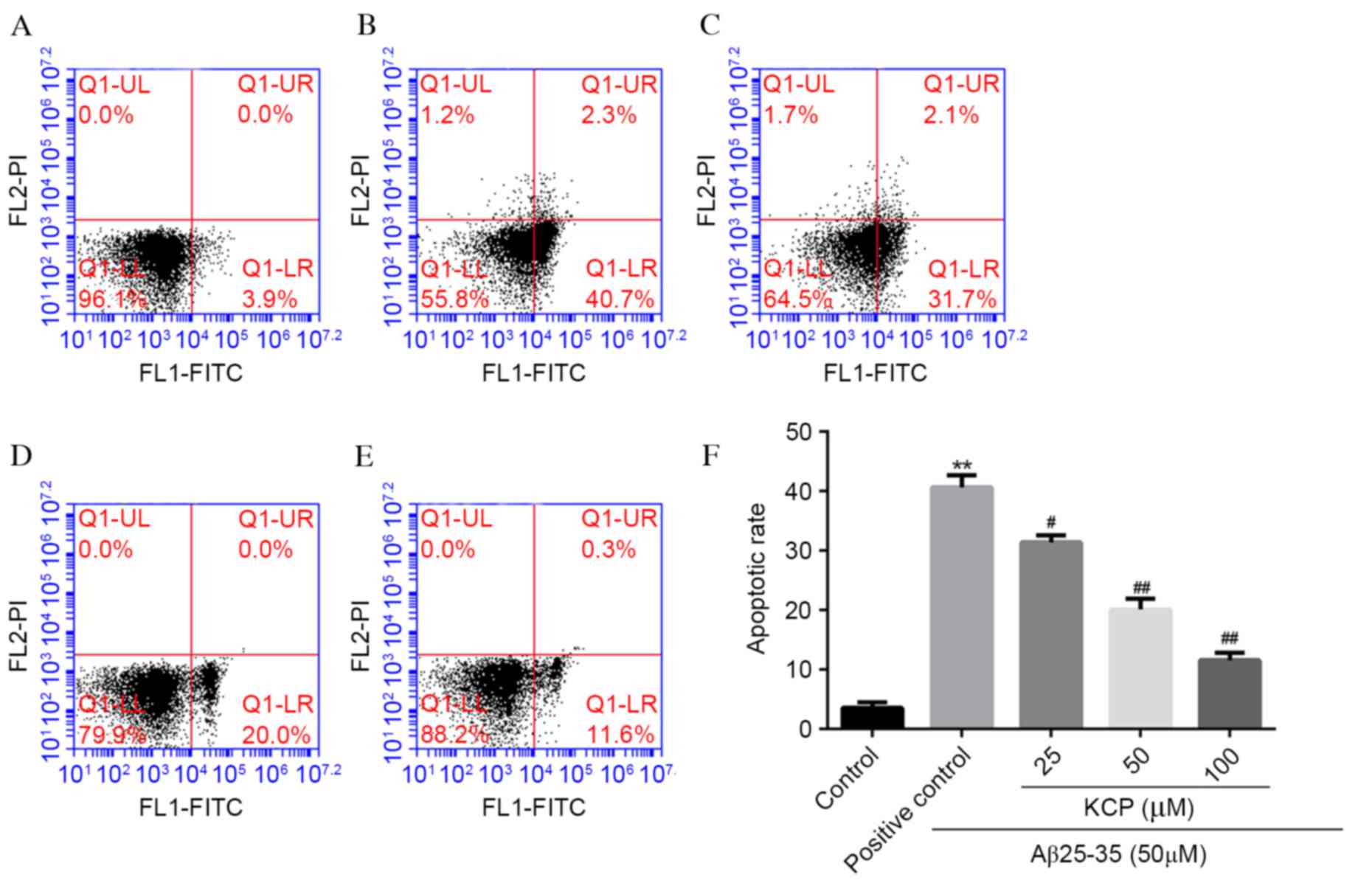
KCP reduces apoptosis in SH-SY5Y
cells
To examine whether Aβ25–35-induced cell
death was the result of apoptosis, flow cytometry was performed.
Cells were stained with annexin V-FITC and PI and apoptotic cells
were defined as annexin V+PI−. In the
untreated group, the apoptotic rate was 3.57±0.95%, which increased
to 40.63±2.0% (P<0.0001; Fig.
3) on exposure to 50 µM Aβ25–35. Following
pretreatment of SH-SY5Y cells with various concentrations of KCP,
the apoptotic rate decreased to 31.40±1.18% (P=0.0023) at 25 µM,
20.00±1.85% (P=0.0002) at 50 µM and 11.57±1.25% (P<0.0001) at
100 µM (Fig. 3). Therefore, KCP
significantly reduced the rate of apoptosis induced by
Aβ25–35, in a dose-dependent manner.
Evaluation of cleaved caspase 3
protein expression levels in SH-SY5Y cells treated with
Aβ25–35 in the absence or presence of KCP
Cleaved caspase 3 is an important marker of
apoptosis; therefore its protein expression levels in SH-SY5Y cells
were analyzed by western blotting (Fig. 4A). SH-SY5Y cells treated with
Aβ25–35 (50 µM) demonstrated increased protein
expression levels of cleaved caspase 3 (P<0.0001; Fig. 4A), indicating activation of
apoptosis. However, following pretreatment with KCP, the protein
expression levels of cleaved caspase 3 decreased significantly in a
dose-dependent manner (P=0.0193, P=0.0002 and P<0.0001,
respectively; Fig. 4A). These
results were verified by immunofluorescence (Fig. 4B).
Role of the JNK signaling pathway in
the protective effects of KCP against Aβ25–35-induced
apoptosis in SH-SY5Y cells
The JNK signaling pathway mediates cellular
apoptosis. In contrast to the untreated group, cells treated with
50 µM Aβ25–35 demonstrated considerably increased p-JNK
protein expression levels (P<0.0001; Fig. 5B). However, treatment of cells with
a combination of 50 µM Aβ25–35 and 25, 50 or 100 µM KCP
significantly decreased p-JNK protein expression levels in a
dose-dependent manner (P=0.0019, P<0.0001 and P<0.0001,
respectively; Fig. 5B) compared
with cells treated with 50 µM Aβ25–35 alone.
Discussion
The present study demonstrated the protective effect
of KCP against Aβ25–35-induced damage. Activation of
caspase 3 and the JNK signaling pathway may be involved in this
process. These findings indicated the neuroprotective potential of
KCP by inhibiting the excessive activation of certain signaling
pathways in neuronal cells.
Apoptotic neuronal cell death is a primary
characteristic of numerous degenerative diseases, including AD.
Caspases, a family of cysteine proteases, are critical for
apoptosis in the central nervous system (19). Caspase 3, which is known as the
final effector of apoptosis, splits into the two subunits (17 and
19 kD) of cleaved caspase 3, and promotes apoptosis by stimulating
certain apoptotic factors (20).
The results of annexin V/PI staining and the protein expression
levels of cleaved caspase 3 supported the finding that KCP may
protect SH-SY5Y neuronal cells from Aβ25–35-induced
apoptosis.
JNK is a serine/threonine kinase that is involved in
cellular responses including proliferation, mitogenic stimuli,
environmental stress and apoptosis (21). The expression and activation of JNK
was first observed in the brains of patients with AD (22). In a subsequent study, JNK
activation was observed in the primary cortical neuron cultures
incubated with Aβ peptides and in transgenic mice overexpressing
mutant PS1 (M146L), as well as in the human AD brain. In addition,
incubation of Aβ peptides with primary cortical neuron cultures
induced JNK activation and cell death (16). Furthermore, the activation of the
JNK signaling pathway has been observed in the
Aβ25–35-induced rat model of AD (23). Inhibiting the activation of the JNK
signaling pathway attenuates the Aβ25–35-induced
toxicity in primary neurons (24).
To examine the importance of the JNK signaling pathway, a previous
study used a JNK inhibitor, which improved learning and long-term
memory in Aβ-injected rats (25).
The JNK inhibitor, CEP-1347 (KT7515) protected PC12 cells and
sympathetic neurons from Aβ-induced death, which indicates that the
JNK signaling pathway acts relatively proximally and triggers the
death mechanism (26). The present
study confirmed that Aβ25–35 activated the JNK signaling
pathway; KCP inhibited the increased activation of this pathway,
thereby inhibiting apoptosis. Although activation of the JNK
signaling pathway appears necessary for Aβ25–35-induced
cell death, it may be one of many possible mechanisms by which
apoptosis is activated in these cells.
The present study has certain limitations, in that
it focuses on the occurrence of apoptosis. It did not verify the
specific process by which KCP attenuates Aβ25–35-induced
apoptosis. Further studies are required to investigate this.
However, the results of the present study suggested that KCP
inhibits Aβ25–35-induced apoptosis of SH-SY5Y cells and
that the JNK signaling pathway is involved in this process.
In conclusion, the results of the present study
demonstrated a preliminary underlying mechanism to support the
hypothesis that KCP possesses neuroprotective properties, and
elucidates the specific role of JNK in this process. The
attenuation of Aβ25–35-induced neuroblastoma cell
cytotoxicity by KCP suggested that KCP may be a potential
therapeutic agent for the treatment of AD.
Acknowledgements
The present study was supported by Chongqing
Municipal Health Bureau (grant no. 20141007).
Glossary
Abbreviations
Abbreviations:
|
Aβ
|
β-amyloid
|
|
AD
|
Alzheimer's disease
|
|
KCP
|
κ-carrageenan pentasaccharide
|
|
JNK
|
c-Jun N-terminal kinase
|
References
|
1
|
Mayeux R and Stern Y: Epidemiology of
Alzheimer disease. Cold Spring Harb Perspect Med.
2:a0062392012.PubMed/NCBI
|
|
2
|
Yao M, Nguyen TV and Pike CJ:
Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal
kinase-dependent downregulation of Bcl-w. J Neurosci. 25:1149–1158.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hardy JA and Higgins GA: Alzheimer's
disease: The amyloid cascade hypothesis. Science. 256:184–185.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tayeb HO, Murray ED, Price BH and Tarazi
FI: Bapineuzumab and solanezumab for Alzheimer's disease: Is the
‘amyloid cascade hypothesis’ still alive? Expert Opin Biol Ther.
13:1075–1084. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Doody RS, Thomas RG, Farlow M, Iwatsubo T,
Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, et al:
Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's
disease. N Engl J Med. 370:311–321. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salloway S, Sperling R, Fox NC, Blennow K,
Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S,
et al: Two phase 3 trials of bapineuzumab in mild-to-moderate
Alzheimer's disease. N Engl J Med. 370:322–333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karran E and Hardy J: Antiamyloid therapy
for Alzheimer's dis-ease-are we on the right road? N Engl J Med.
370:377–378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Golde TE, Schneider LS and Koo EH: Anti-aβ
therapeutics in Alzheimer's disease: The need for a paradigm shift.
Neuron. 69:203–213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaminsky YG, Marlatt MW, Smith MA and
Kosenko EA: Subcellular and metabolic examination of amyloid-beta
peptides in Alzheimer disease pathogenesis: Evidence for Abeta
(25–35). Exp Neurol. 221:26–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan H, Zhang W, Li X, Lü X, Li N, Gao X
and Song J: Preparation and in vitro antioxidant activity of
kappa-carrageenan oligosaccharides and their oversulfated,
acetylated, and phosphorylated derivatives. Carbohydr Res.
340:685–692. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klarzynski O, Descamps V, Plesse B, Yvin
JC, Kloareg B and Fritig B: Sulfated fucan oligosaccharides elicit
defense responses in tobacco and local and systemic resistance
against tobacco mosaic virus. Mol Plant Microbe Interact.
16:115–122. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guven KC, Ozsoy Y and Ulutin ON:
Anticoagulant, fibrinolytic and antiaggregant activity of
carrageenans and alginic acid. Botanica Marina. 34:429–432.
1991.
|
|
13
|
McGeer PL: Immune mechanisms in
neurodegeneration. Drugs Today. 32:149–158. 1996.
|
|
14
|
Xu L, Yao Z, Wu H, Wang F and Zhang S: The
immune regulation of κ-carrangeenan oligosaccharide and its
desulfated derivatives on LPS-activated microgial cells.
Neurochemistry International. 61:689–696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ip YT and Davis RJ: Signal transduction by
the c-Jun N-terminal kinase (JNK)-from inflammation to development.
Curr Opin Cell Biol. 10:205–219. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shoji M, Iwakami N, Takeuchi S, Waragai M,
Suzuki M, Kanazawa I, Lippa CF, Ono S and Okazawa H: JNK activation
is associated with intracellular beta-amyloid accumulation. Brain
Res Mol Brain Res. 85:221–233. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu X, Rottkamp CA, Kubat Z, et al:
Amyloid-beta toxicity is metal-mediated and acts through the
activation of JNk/SAPK pathway in Alzheimer disease. Society for
Neuroscience Abstracts. 27:8552001.
|
|
18
|
Fang F and Liu GT: Novel squamosamide
derivative (compound FLZ) attenuates Abeta25-35-induced toxicity in
SH-SY5Y cells. Acta Pharmacol Sin. 29:152–160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raynaud F and Marcilhac A: Implication of
calpain in neuronal apoptosis. A possible regulation of Alzheimer's
disease. FEBS J. 273:3437–3443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boatright KM and Salvesen GS: Mechanisms
of caspase activation. Curr Opin Cell Biol. 15:725–731. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen YR, Wang X, Templeton D, Davis RJ and
Tan TH: The role of c-Jun N-terminal kinase (JNK) in apoptosis
induced by ultraviolet C and gamma radiation. Duration of JNK
activation may determine cell death and proliferation. J Biol Chem.
271:31929–31936. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kumagae Y, Zhang Y, Kim OJ and Miller CA:
Human c-Jun N-terminal kinase expression and activation in the
nervous system. Brain Res Mol Brain Res. 67:10–17. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ghasemi R, Zarifkar A, Rastegar K,
Maghsoudi N and Moosavi M: Repeated intra-hippocampal injection of
beta-amyloid 25–35 induces a reproducible impairment of learning
and memory: Considering caspase-3 and MAPKs activity. Eur J
Pharmacol. 726:33–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qin Y, Chen Z, Han X, Wu H, Yu Y, Wu J,
Liu S and Hou Y: Progesterone attenuates Aβ(25–35)-induced neuronal
toxicity via JNK inactivation and progesterone receptor membrane
component 1-dependent inhibition of mitochondrial apoptotic
pathway. J Steroid Biochem Mol Biol. 154:302–311. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yenki P, Khodagholi F and Shaerzadeh F:
Inhibition of phosphorylation of JNK suppresses Aβ-induced ER
stress and upregulates prosurvival mitochondrial proteins in rat
hippocampus. J Mol Neurosci. 49:262–269. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Troy CM, Rabacchi SA, Xu Z, Maroney AC,
Connors TJ, Shelanski ML and Greene LA: Beta-Amyloid-induced
neuronal apoptosis requires c-Jun N-terminal kinase activation. J
Neurochem. 77:157–164. 2001. View Article : Google Scholar : PubMed/NCBI
|