Introduction
Joubert syndrome (JS) is a recessive or X-linked
genetic disorder, which is characterized by hypotonia,
apnea/hyperpnea in infancy, oculomotor apraxia, psychomotor
delay/mental retardation, and cerebellar vermis hypoplasia and
dysplasia. JS is accompanied by brainstem abnormalities, which
result in radiological detection of the ‘molar tooth sign’
(1–6). JS can be classified into six
phenotypic subtypes, based on other clinical abnormalities detected
alongside JS, including occipital encephalocele, polymicrogyria,
kidney lesions, polydactyly, hepatic fibrosis and ocular coloboma
(6). JS has an incidence of 1 in
80,000–100,000 individuals in the United States (6). Several of the JS phenotypes are
similar to those associated with Meckel syndrome (MKS) (7–9),
which is a lethal recessive disorder that is characterized by renal
cystic dysplasia; occipital encephalocele, or other central nervous
system phenotypes; polydactyly and hepatic fibrosis. Therefore,
some researchers consider JS and MKS to be different phenotypes of
the same disease. JS is clinically and genetically heterogeneous,
and overlaps with several other ciliopathies, including
nephronophthisis, Senior-Loken syndrome and MKS (10). To date, >30 causative genes
(11,12) have been identified in JS.
Coiled-coil and C2 domain containing 2A
(CC2D2A) was initially described in JS and MKS in 2008, and
further investigations have elucidated the role of CC2D2A in
ciliary function (13–19). Previous studies have linked JS, and
other syndromic disorders, to defective cilia (14,16,20–22).
The present study identified a JS family with novel compound
heterozygous mutations in CC2D2A.
Case report
Ethical permission for genetic analysis and
collection of test data in the present study was granted by the
Research Ethics Committee at Zunyi Medical University (Zunyi,
China). Written informed consent was obtained from the participants
or participants' family. The proband was an 8-month-old male who
was diagnosed with JS before being subjected to cerebral magnetic
resonance imaging (MRI) using a 1.5 Tesla MRI scanner (Siemens AG,
Munich, Germany). The patient matched the JS diagnotic criteria:
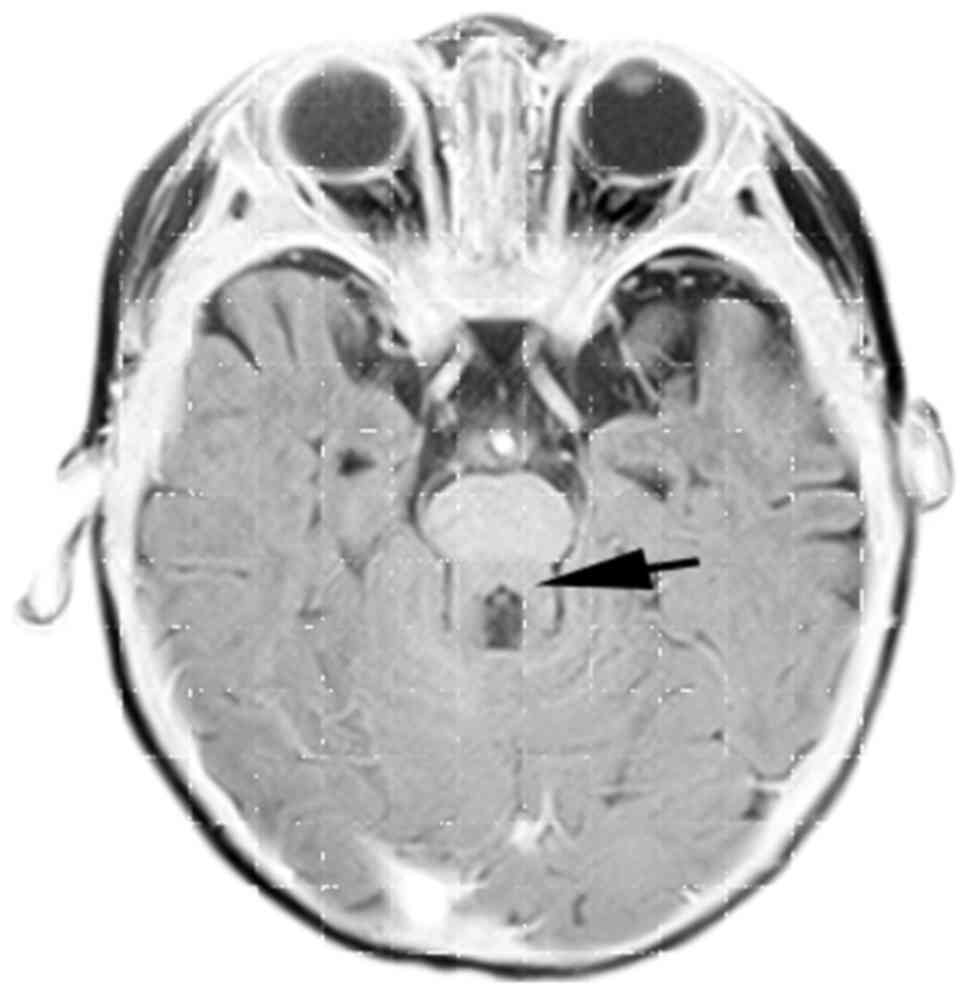
Cranial MRI demonstrated brainstem ‘molar tooth sign’ (Fig. 1) (6,23).
Further examinations indicated that the proband exhibited
hypotonia, psychomotor delay/intellectual disability, nystagmus and
jerky eye movements. Other tests, including complete blood count,
blood biochemistry, serum lactate, urine ammonia, plasma and urine
amino acids, and metabolic screening of blood, were conducted in
our laboratory.
Upon JS diagnosis, 10 µg DNA was extracted from a
blood sample collected from the proband according to the protocol
of the Invitrogen genomic DNA extraction kit (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), in order to determine
what genetic alterations caused the disease. High-throughput exome
sequencing was performed on the DNA samples obtained from the
proband by Shenzhen BGI Diagnosis Technology Co., Ltd. (Shenzhen,
China). The sequencing results revealed that the proband possessed
CC2D2A compound heterozygous mutations
(c.2581G>A/c.2848C>T), and demonstrated that two
CC2D2A alleles were mutated (Fig. 2). The c.2581G>A mutation is
located on exon 21, whereas the c.2848C>T alteration is situated
on exon 23 of the gene. The c.2581G>A mutation results in an
amino acid replacement (p.Asp861Asn), whereas the c.2848C>T
mutation leads to an immediate stop code (p.Arg950Ter), which
results in a truncated form of the CC2D2A protein. To
determine the source of the mutations, the proband's parents and
grandparents were recruited for genetic examination. Furthermore,
the mother was pregnant (6 months) and also decided to screen the
fetus. An MRI analysis indicated that the female fetus displayed
molar tooth sign. Blood samples were collected for DNA extraction
from the proband's parents, grandparents, and the proband. The
present study screened for mutations in the samples obtained from
the proband's available parents and grandparents using routine
polymerase chain reaction (PCR) and DNA sequencing on exons 21 and
23. The PCR primers were designed and synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.). PCR was performed according to
the manufacturer's protocol [Thermo Scientific Dream Taq Green PCR
Master Mix (2X); #K1081; Thermo Fisher Scientific, Inc.]. Briefly,
50 pg patient DNA was mixed with 1 µM PCR primers and 25 µl Taq
Green PCR Master Mix; nuclease-free water was added to ensure the
final volume was 50 µl. The mixture was initially denatured for 3
min at 95°C; PCR was then conducted for 35 cycles at 95°C for 30
sec, 58°C for 30 sec and 72°C for 1 min; finally, the PCR products
underwent extension at 72°C for 5 min. DNA sequencing was carried
out by Shenzhen BGI Diagnosis Technology Co., Ltd. The PCR primer
sequences were as follows: Exon 21, forward
5′-GCACTTTTATGCACTGGACAGA-3′, reverse 5′-CAGCACCCATGTTCTTAAACAG-3′
(496 bp); exon 23, forward 5′-TGTGGCCTCTATTTCTTCCTGA-3′; reverse,
5′-GCTAAGTGGCTTGCATAAGCTC-3′ (470 bp). PCR sequencing indicated
that both parents were CC2D2A mutation heterozygotes: The
father carried the c.2581G>A mutation, whereas the mother
carried the c.2848C>T mutation (Fig. 3). The proband's parents displayed
normal phenotypes due to the heterogeneity of the mutations. In
addition, the proband's maternal grandmother was also a carrier of
the c.2848C>T mutation.
The mother's pregnancy was terminated following
detection of the molar tooth sign in utero. Fetal brain
tissue was collected post-termination for genetic and histological
analysis. Similar to the proband, the female fetus was demonstrated
to possess the CC2D2A compound heterozygous mutations
through PCR and DNA sequencing using post-termination brain tissue.
With the exception of the molar tooth sign, no observable
abnormalities were detected in the fetal cerebellar vermis, based
on routine hematoxylin-eosin staining, probably due to the young
age of the fetus (data not shown). In some cases, JS is accompanied
with defects in other organs, such as the kidney and liver. The
present study conducted histological and immunohistochemical (IHC)
analyses using CC2D2A antibody (cat. no. 22293-1-AP; ProteinTech
Group, Inc., Rosemont, IL, USA) on these fetal tissues
post-termination. Briefly, collected tissues were fixed in 4%
paraformaldehyde for 24 h. The tissues were then embedded in
paraffin, according to the embedding machine manufacturer's
protocol. Each paraffin block was cut into 5 µm sections for
hematoxylin-eosin (H&E) staining and IHC analyses. Paraffin
sections were deparaffinized and hydrated using xylene and a graded
series of alcohol. For H&E staining, the tissue sections were
stained with 0.5% hematoxylin and 0.5% eosin. For IHC, tissue
section antigen retrieval was conducted using a citrate-based
Antigen Unmasking Solution (cat. no. H-3300; Vector Laboratories,
Inc., Burlingame, CA, USA). IHC was conducted using the Vectastain
Universal Elite ABC kit (cat. no. PK-6200; Vector Laboratories,
Inc.) according to the manufacturer's protocol. CC2D2A antibody was
used at a 1:200 dilution, and tissues were incubated with it for 30
min at room temperature, following antigen retrieval and blocking
with normal horse serum for 1 h. Subsequently, tissue sections were
incubated with diluted anti-rabbit biotinylated secondary antibody
(provided in the ABC kit) for 30 min. All the operations were
performed at room temperature. In the fetal kidney samples, besides
increased chronic inflammation and abnormal glomeruli, no renal
cysts were observed. In addition, hepatic fibrosis was not observed
in the affected fetal liver samples. Renal and hepatic involvement
in JS is a progressive condition, which can be completely
asymptomatic for several years; therefore, renal cysts/hepatic
fibrosis may not appear at all while the patients develop this
progressive disease. Therefore, in the fetus, it is unsurprising
that no JS-related defects were observed in the kidney and liver at
the age of 6 months.
As aforementioned, routine IHC analysis was used to
examine CC2D2A protein expression in these tissues, and all tissues
were found to be CC2D2A positive, thus indicating that
abnormal/truncated CC2D2A proteins are produced by mutated
CC2D2A alleles (data not shown).
Discussion
The present study identified a JS family with novel
compound heterozygous mutations in CC2D2A. Both mutations
detected are not present in the 1000 Genomes database (1000genomes.org) or the NHLBI Exome Sequencing Project
(http://evs.gs.washington.edu/EVS/),
thus indicating that they are novel CC2D2A mutations. Since
the proband's parents are mutation carriers and not affected, both
mutations are causative based on the recessive genetic two-hit
theory.
In the present family, JS was caused by novel
compound heterozygous mutations in CC2D2A inherited from the
parents. These findings may be used to establish prenatal molecular
diagnostic criteria, and may be beneficial in future pregnancies.
In the present study, the proband's mother was pregnant and was
subject to prenatal diagnosis based on the family history. The
pregnancy was terminated at the parents' choice due to the
confirmed compound heterozygous CC2D2A mutations and
phenotypic defects.
Acknowledgements
The present authors would like to thank Ms. Sue
Schoen (University of Rochester Medical Center) for reviewing the
present study. The present study was supported by the Guizhou
Science and Technology Foundation [LKZ (2012) 39].
References
|
1
|
Joubert M, Eisenring JJ and Andermann F:
Familial dysgenesis of the vermis: A syndrome of hyperventilation,
abnormal eye movements and retardation. Neurology. 18:302–303.
1968.
|
|
2
|
Patel S and Barkovich AJ: Analysis and
classification of cerebellar malformations. AJNR Am J Neuroradiol.
23:1074–1087. 2002.PubMed/NCBI
|
|
3
|
Valente EM, Dallapiccola B and Bertini E:
Joubert syndrome and related disorders. Handb Clin Neurol.
113:1879–1888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Poretti A, Boltshauser E and Valente EM:
The molar tooth sign is pathognomonic for Joubert syndrome! Pediatr
Neurol. 50:e15–e16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parisi MA: Clinical and molecular features
of Joubert syndrome and related disorders. Am J Med Genet C Semin
Med Genet 151C. 326–340. 2009.
|
|
6
|
Romani M, Micalizzi A and Valente EM:
Joubert syndrome: Congenital cerebellar ataxia with the molar
tooth. Lancet Neurol. 12:894–905. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salonen R and Paavola P: Meckel syndrome.
J Med Genet. 35:497–501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kyttälä M, Tallila J, Salonen R, Kopra O,
Kohlschmidt N, Paavola-Sakki P, Peltonen L and Kestilä M: MKS1,
encoding a component of the flagellar apparatus basal body
proteome, is mutated in Meckel syndrome. Nat Genet. 38:155–157.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Szymanska K, Hartill VL and Johnson CA:
Unraveling the genetics of Joubert and Meckel-Gruber syndromes. J
Pediatr Genet. 3:65–78. 2014.PubMed/NCBI
|
|
10
|
Otto EA, Ramaswami G, Janssen S, Chaki M,
Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT, et al:
Mutation analysis of 18 nephronophthisis associated ciliopathy
disease genes using a DNA pooling and next generation sequencing
strategy. J Med Genet. 48:105–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kroes HY, Monroe GR, van der Zwaag B,
Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijman IJ,
Kloosterman WP, Giles RH, et al: Joubert syndrome: Genotyping a
Northern European patient cohort. Eur J Hum Genet. 24:214–220.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsurusaki Y, Kobayashi Y, Hisano M, Ito S,
Doi H, Nakashima M, Saitsu H, Matsumoto N and Miyake N: The
diagnostic utility of exome sequencing in Joubert syndrome and
related disorders. J Hum Genet. 58:113–115. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Noor A, Windpassinger C, Patel M,
Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F,
Paterson AD, et al: CC2D2A, encoding a coiled-coil and C2 domain
protein, causes autosomal-recessive mental retardation with
retinitis pigmentosa. Am J Hum Genet. 82:1011–1018. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tallila J, Jakkula E, Peltonen L, Salonen
R and Kestilä M: Identification of CC2D2A as a Meckel syndrome gene
adds an important piece to the ciliopathy puzzle. Am J Hum Genet.
82:1361–1367. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jones D, Fiozzo F, Waters B, McKnight D
and Brown S: First-trimester diagnosis of Meckel-Gruber syndrome by
fetal ultrasound with molecular identification of CC2D2A mutations
by next-generation sequencing. Ultrasound Obstet Gynecol.
44:719–721. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gorden NT, Arts HH, Parisi MA, Coene KL,
Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen
D, et al: CC2D2A is mutated in Joubert syndrome and interacts with
the ciliopathy-associated basal body protein CEP290. Am J Hum
Genet. 83:559–571. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Williams CL, Li C, Kida K, Inglis PN,
Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et
al: MKS and NPHP modules cooperate to establish basal
body/transition zone membrane associations and ciliary gate
function during ciliogenesis. J Cell Biol. 192:1023–1041. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mougou-Zerelli S, Thomas S, Szenker E,
Audollent S, Elkhartoufi N, Babarit C, Romano S, Salomon R, Amiel
J, Esculpavit C, et al: CC2D2A mutations in Meckel and Joubert
syndromes indicate a genotype-phenotype correlation. Hum Mutat.
30:1574–1582. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Veleri S, Manjunath SH, Fariss RN,
May-Simera H, Brooks M, Foskett TA, Gao C, Longo TA, Liu P,
Nagashima K, et al: Ciliopathy-associated gene Cc2d2a promotes
assembly of subdistal appendages on the mother centriole during
cilia biogenesis. Nat Commun. 5:42072014.PubMed/NCBI
|
|
20
|
Iannicelli M, Brancati F, Mougou-Zerelli
S, Mazzotta A, Thomas S, Elkhartoufi N, Travaglini L, Gomes C,
Ardissino GL, Bertini E, et al: Novel TMEM67 mutations and
genotype-phenotype correlates in meckelin-related ciliopathies. Hum
Mutat. 31:E1319–E1331. 2010.PubMed/NCBI
|
|
21
|
Stephen LA, Tawamie H, Davis GM, Tebbe L,
Nürnberg P, Nürnberg G, Thiele H, Thoenes M, Boltshauser E, Uebe S,
et al: TALPID3 controls centrosome and cell polarity and the human
ortholog KIAA0586 is mutated in Joubert syndrome (JBTS23). Elife.
4(pii): e080772015.PubMed/NCBI
|
|
22
|
Doherty D: Joubert syndrome: Insights into
brain development, cilium biology, and complex disease. Semin
Pediatr Neurol. 16:143–154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maria BL, Hoang KB, Tusa RJ, Mancuso AA,
Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl
DM, et al: ‘Joubert syndrome’ revisited: Key ocular motor signs
with magnetic resonance imaging correlation. J Child Neurol.
12:423–430. 1997. View Article : Google Scholar : PubMed/NCBI
|