Introduction
Lung cancer is a common malignancy and the leading
cause of cancer-associated mortality worldwide (1). Non-small cell lung cancer (NSCLC)
accounts for ~80% of lung cancers (2). Despite tremendous progress being made
in surgical techniques, chemotherapeutic agents, radiotherapy and
novel molecular targeted drugs in previous decades, the prognosis
of NSCLC remains poor, making the development of comprehensive
treatments for the disease an urgent requirement. Oridonin,
alternately known as guidongnin, is a tetracyclic diterpenoid
compound extracted and purified from a traditional Chinese herb,
Rabdosia rubescens, which is a member of the Salvia
family. Previous studies have demonstrated that oridonin exhibits
cytotoxicity, strong anti-tumor activity and is a powerful tumor
suppressor (3–5). Oridonin directly inhibits the growth
of >20 human cancer cell lines, including NB4 leukemia cells,
A549 lung adenocarcinoma cells, HeLa cervical carcinoma cells,
A357-S2 melanoma cells and SGC-7901 gastric cancer cells (6–10).
G2/M cell cycle arrest occurs when DNA is damaged,
preventing cells from entering mitosis and providing an opportunity
for DNA repair in order to maintain genomic stability (11–14).
The ataxia telangiectasia mutated (ATM) gene encodes a
serine/threonine protein kinase associated with DNA repair in an
array of tumors (12,13). As the product of the ATM gene, ATM
possesses the property of autophosphorylation and is of vital
significance to the DNA damage checkpoint, the repair of damaged
DNA and apoptosis (11). Along
with other proteins including p53 and checkpoint kinase (CHK2), ATM
is involved in cell cycle arrest in response to situations where
DNA is damaged (15).
The present study aimed to investigate whether cell
cycle arrest contributes to oridonin-induced inhibition of
proliferation, and attempted to explore the molecular mechanisms
underlying this in A549 cells.
Materials and methods
Reagents and antibodies
Oridonin (≥98%) was purchased from Shanghai Standard
Technology Co., Ltd. (Shanghai, China) and dissolved in dimethyl
sulfoxide (DMSO; final concentration ≤0.1%). Dulbecco's modified
Eagle's medium (DMEM) and calf serum were obtained from Gibco;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Antibodies of
H2A histone family member X (H2AX; 1:100; cat. no. 7631), γ-H2AX
(1:100 cat. no. 2577), ATM serine/threonine kinase (ATM; 1:500;
cat. no. 2873), p-p53 (S15; 1:1,000; cat. no. 9286), checkpoint
kinase 2 (CHK2; 1:1,000; cat. no. 3340), and p-CHK2 (T68; 1:500;
cat. no. 2661) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). ATM pS1981 (1:100, cat. no. 200-301-400) was
purchased from Rockland, Inc. (Limerick, PA, USA). Secondary
antibodies goat anti-rabbit (1:5,000; cat. no. sc-2030) and goat
anti-mouse (1:5,000; cat. no. sc-2031.) were labeled with
horseradish peroxidase. β-actin (cat. no. sc-47778; 1:1,000), lamin
B (cat. no. sc-6217; 1:1,000) and p53 (cat. no. sc-6243; 1:200)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). RNase and propidium iodide (PI) solution were
purchased from Sigma-Aldrich; Merck Millipore (Darmstadt,
Germany).
Cell culture
Human A549 lung cancer cells were purchased from
China Center for Type Culture Collection of Wuhan University
(Wuhan, China). Cells were cultured in DMEM supplemented with 10%
calf serum and incubated in a humidified atmosphere (5%
CO2) at 37°C. Cells were passaged routinely and
logarithmically growing cells were used in the experiments as
needed.
Flow cytometry
A549 cells (~1.0×104) were treated for 48
h with DMSO or oridonin at different concentrations (16, 32 and 64
µmol/l), collected and washed with phosphate-buffered saline (PBS)
twice, followed by fixation with 70% ice-cold alcohol at 4°C for a
least 1 h. Cells were rinsed in ice-cold PBS twice, then suspended
in 3 ml PBS containing ribonuclease (Sigma-Aldrich; Merck
Millipore) with the final concentration of 100 µg/l. The cells were
then incubated in a 37°C water-bath for 30 min and stained with 50
µg/l PI for 30 min in the dark. The percentages of cells at
different phases of the cell cycle were detected by flow cytometry
(Cytoflex, Beckman Coulter, Inc., Brea, CA, USA), with absorbance
measured at 488 nm (CytExpert, Beckman Coulter, Inc.).
Western blot
Oridonin or DMSO treated A549 cells were collected
to a final concentration of 2×106 cells/ml. Proteins
were extracted using a Protein Extraction Solution kit purchased
from Beijing SBS Genetech Co., Ltd. (Beijing, China). Protein
concentration was determined by bicinchoninic acid assay. Proteins
were denatured in the presence of an equal volume of 2X sample
buffer by boiling for 10 min at 95°C prior to SDS-PAGE. Following
gel electrophoresis, the protein (50 µg per lane) was transferred
onto a polyvinylidene difluoride membrane at 260 mA for 90 min. The
membrane was then blocked with TBS containing 5% bovine serum
albumin (BSA, Thermo Fisher Scientific, Inc.), followed by
incubation with primary antibody overnight at 4°C. The membrane was
subsequently washed four times with TBST (TBS plus 0.1% Tween-20)
for 15 min, and incubated with secondary antibody (1:1,000) for 1 h
at room temperature. The membrane was then washed four times again
in TBST for 15 min and the bound antibodies were detected using
horseradish peroxidase-electrochemiluminescence. β-actin was used
as a loading control. The Western blot results were repeated three
times.
Immunofluorescence
Cells (~1×104) were fixed with
pre-chilled (−20°C) acetone-methanol for 15 min and then blocked
with 5% BSA at room temperature for 1 h. γ-H2AX and p-ATM (S1981)
antibodies were added to the slides and incubated at 4°C overnight.
Following washing, specimens were incubated with secondary
antibodies for 1 h at room temperature. The cells were then covered
with mounting medium containing 4′,6-diamidino-2-phenylindole.
Images were taken using SPOT 5.0 Advanced software (SPOT Imaging)
with a Nikon TE1000 wide-field microscope system (Nikon).
Statistical analysis
Data were analyzed with SPSS 10.0 software (SPSS,
Inc., Chicago, IL, USA) and are expressed as the mean ± standard
deviation if not otherwise indicated. Student's t-test was
performed. P<0.05 was considered to indicate a statistically
significant difference.
Results
Oridonin induces cell cycle arrest at
G2/M phase in A549 cells
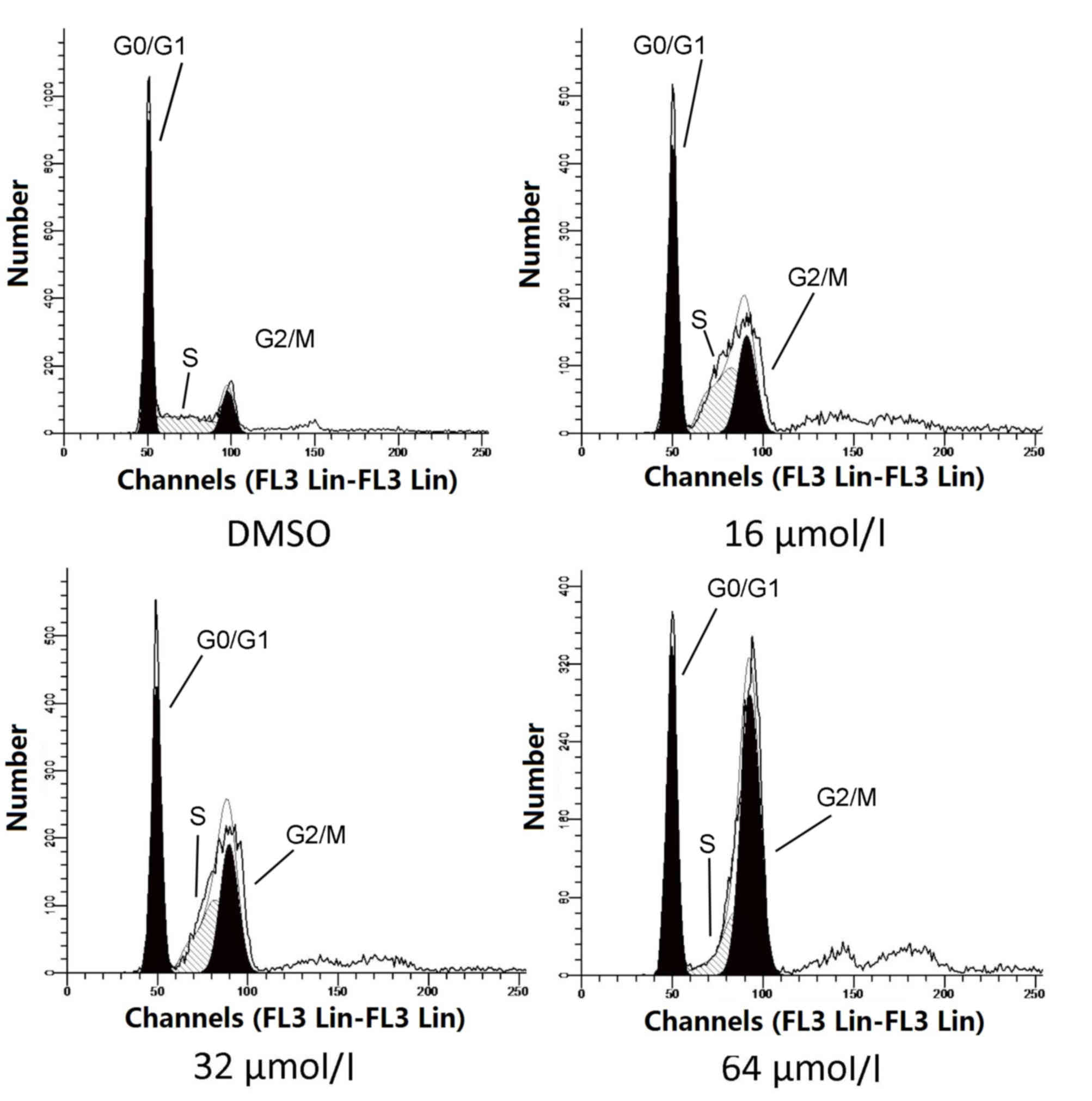
To determine whether oridonin-induced cell cycle
arrest contributes to tumor growth inhibition, A549 cells were
exposed to oridonin at different concentrations (16, 32 and 64
µmol/l) for 48 h, and same volume of DMSO was administered as a
vehicle control. Cells were subsequently harvested and cell cycle
activity assessed by flow cytometry (Fig. 1). Quantitative analysis revealed
that the proportion of G2/M phase cells was significantly
increased, dose-dependently, in oridonin-treated A549 cells treated
with 16 (P=0.014; Table I), 32
(P=0.009; Table I) and 64 µmol/l
oridonin (P=0.00003; Table I)
compared with DMSO-treated cells. By contrast, the proportion of
G0/G1 phase cells decreased as a result of oridonin treatment
compared with DMSO treatment (Table
I). These results demonstrated that oridonin arrests A549 cells
at the G2/M phase of the cell cycle in a dose-dependent manner.
 | Table I.Effect of oridonin treatment on the
cell cycle of A549 cells. |
Table I.
Effect of oridonin treatment on the
cell cycle of A549 cells.
| Group | G1 (%) | S (%) | G2/M (%) |
|---|
| DMSO | 70.10±0.6 | 19.73±0.8 | 10.17±1.1 |
| 16 µmol/l | 51.72±4.9 | 31.24±3.89 |
17.04±3.34a |
| 32 µmol/l | 46.68±5.1 | 26.3±4.71 |
27.02±4.43a |
| 64 µmol/l | 34.7±5.2 | 17.23±4.89 |
48.07±5.89a |
Oridonin induces G2/M arrest by
facilitating ATM activation in A549 cells
To investigate whether cell cycle-associated
proteins (γ-H2AX, H2AX, lamin-B, p-ATM (S1981), ATM, p-CHK2 (T68),
CHK2, p-p53 and p53) are involved in the oridonin-induced G2/M
arrest in A549 cells, western blot was performed to evaluate the
level of each protein. Following treatment with 16, 32 and 64
µmol/l oridonin, p-ATM (S1981), p-CHK2 (T68), p-p53, p53 and γ-H2AX
protein levels in A549 cells increased (Fig. 2). The higher the concentration of
oridonin used, the greater upregulation of p-ATM (S1981), p-CHK2
(T68), p-p53, p53 and γ-H2AX protein expression levels was
presented (Fig. 2).
Effects of oridonin on p-ATM (S1981)
and γ-H2AX
To further explore the function of p-ATM (S1981) and
γ-H2AX on cell cycle arrest, A549 cells were treated with 16, 32
and 64 µmol/l oridonin and immunostaining was performed to
investigate changes in expression of these two proteins. Both p-ATM
(S1981) and γ-H2AX were upregulated following oridonin treatment
(Fig. 3). Both p-ATM (S1981) and
γ-H2AX were localized in the cell nucleus (Fig. 3).
Discussion
Lung cancer is one of the most common malignant
tumors, and is associated with poor overall survival, therefore
research into effective treatments is required (16). Previous in vitro studies
have explored the anti-tumor effects of oridonin, and have
demonstrated that it inhibits abnormal cell proliferation and
induces apoptosis in various human tumor cell lines (6,8,9,17–23).
The cell cycle is a fundamental cellular event,
which is regulated at multiple levels by various factors in
vitro and in vivo. Cell proliferation, division,
apoptosis and necrosis all occur at certain phases in the cell
cycle (24). Cells are programmed
to enter pathological processes if certain phases of the cell cycle
are abnormal (25). It is well
established that cell cycle regulation is associated with the
formation and development of tumors. Uncontrolled cell growth
resulting from loss of cell cycle regulation is a characteristic
common to almost all tumors (26).
The G2/M phase checkpoint is the last opportunity for cell repair
prior to mitosis (15). The
present study demonstrated that the proportion of G2/M phase cells
significantly increased following treatment with 16, 32 and 64
µmol/l oridonin. Therefore, oridonin is able to induce cell cycle
arrest at the G2/M checkpoint in A549 cells, and inhibit cell
growth by interfering with mitotic progression.
DNA is constantly damaged by exogenous and
endogenous factors during the cell cycle. Damaged DNA activates
cell cycle checkpoints and induce cells to repair damaged DNA prior
to entering the mitotic phase, so that genome integrity is
maintained. Failure of DNA repair mechanisms or the accumulation of
DNA damage-associated factors may lead to uncontrolled cell
proliferation, which may eventually result in carcinogenesis. DNA
damage induces phosphorylation of H2AX and activation of ATM and
ATR serine/threonine kinase (ATR), which recognize DNA damage and
transmit signals to downstream targets to promote cell cycle
arrest, repair damaged DNA or induce apoptosis (27–29).
Therefore, ATM and ATR are important for maintaining genomic
stability and the prevention of tumorigenesis. p53 is an important
tumor suppressor, originally identified as a protein bound to the
large T antigen of the SV40 tumor virus (30). Mutations of p53 have been
implicated in multiple human cancers (31). It has previously been demonstrated
that p53 is involved in the DNA damage response and the inhibition
of tumor growth (32). A previous
study also revealed that p53 is involved in oridonin-induced cell
cycle arrest in A549 cells (7).
The present study demonstrated that p-ATM (S1981),
p-CHK2 (T68), p-p53, p53 and γ-H2AX protein levels were all
increased in oridonin-treated cells, indicating that oridonin
induces cell cycle arrest via activation of the ATM pathway.
Damaged DNA induces H2AX protein phosphorylation to γ-H2AX, which
subsequently activates ATM through autophosphorylation of ATM on
Ser1981 (33–35). p-ATM (S1981) subsequently
facilitates the phosphorylation and activation of CHK2 and p53,
leading to cell cycle arrest (11). Oridonin activates proteins
associated with multiple checkpoints, which trigger downstream
cascade reactions and activates the network system regulated by ATM
and ATR (15). With the
participation of associated regulatory proteins, the induced
proteins transfer DNA damage signals to downstream proteins of
signal transduction and act on effector proteins, which may result
in distinct effects including cell cycle arrest, apoptosis, DNA
repair and transcriptional program activation induced through the
ATM-p53-CHK2 pathway (36). As
mentioned previously, DNA damage induces H2AX phosphorylation,
which is recognized as a DNA damage biomarker (37). In the present study,
immunofluorescence staining data confirmed that oridonin treatment
induced DNA damage, with increased levels of γ-H2AX in the nucleus
of treated cells. Increased p-ATM (S1981) in the nucleus following
oridonin treatment demonstrated that DNA damage resulted in
increased levels of activated ATM in the cell nucleus.
To the best of our knowledge, this is the first
report to demonstrate that oridonin-induced A549 cell cycle arrest
occurs via activation of the ATM signaling pathway, although there
are some limitations present. Downstream effector protein
expression levels were not evaluated by western blotting, which
prevented the investigation of detailed mechanisms underlying
oridonin-induced cell cycle arrest in A549 cells. In addition, it
remains unclear if ATM or CHK2 directly induced the activation of
p53. Further experiments are required to address these
questions.
In conclusion, the present study demonstrated that
oridonin induces G2/M cell cycle arrest through activating the ATM
signaling pathway to inhibit proliferation in A549 cells. Future
investigations may determine whether oridonin treatment induces the
apoptosis of A549 cells.
Acknowledgements
The present study was supported by the Shenzhen
Science & Technology Research & Development Project (grant
no: JCYJ20130401112244855).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu Z, Ouyang L, Peng H and Zhang WZ:
Oridonin: Targeting programmed cell death pathways as an
anti-tumour agent. Cell Prolif. 45:499–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu DL, Bu HQ, Jin HM, Zhao JF, Li Y and
Huang H: Enhancement of the effects of gemcitabine against
pancreatic cancer by oridonin via the mitochondrial
caspase-dependent signaling pathway. Mol Med Rep. 10:3027–3034.
2014.PubMed/NCBI
|
|
5
|
Bu HQ, Luo J, Chen H, Zhang JH, Li HH, Guo
HC, Wang ZH and Lin SZ: Oridonin enhances antitumor activity of
gemcitabine in pancreatic cancer through MAPK-p38 signaling
pathway. Int J Oncol. 41:949–958. 2012.PubMed/NCBI
|
|
6
|
Liu J, Huang R, Lin D, Wu X, Peng J, Lin
Q, Pan X, Zhang M, Hou M and Chen F: Apoptotic effect of oridonin
on NB4 cells and its mechanism. Leuk Lymphoma. 46:593–597. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Liu JH, Chai K, Tashiro S, Onodera
S and Ikejima T: Inhibition of c-Met promoted apoptosis, autophagy
and loss of the mitochondrial transmembrane potential in
oridonin-induced A549 lung cancer cells. J Pharm Pharmacol.
65:1622–1642. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu HZ, Yang YB, Xu XD, Shen HW, Shu YM,
Ren Z, Li XM, Shen HM and Zeng HT: Oridonin induces apoptosis via
PI3K/Akt pathway in cervical carcinoma HeLa cell line. Acta
Pharmacol Sin. 28:1819–1826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang CL, Wu LJ, Tashiro S, Onodera S and
Ikejima T: Oridonin induced A375-S2 cell apoptosis via
bax-regulated caspase pathway activation, dependent on the
cytochrome c/caspase-9 apoptosome. J Asian Nat Prod Res. 6:127–138.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao SY, Li J, Qu XY, Zhu N and Ji YB:
Downregulation of Cdk1 and cyclinB1 expression contributes to
oridonin-induced cell cycle arrest at G2/M phase and growth
inhibition in SGC-7901 gastric cancer cells. Asian Pac J Cancer
Prev. 15:6437–6441. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Boohaker RJ and Xu B: The versatile
functions of ATM kinase. Biomed J. 37:3–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan HQ, Huang XB, Ke SZ, Jiang YN, Zhang
YH, Wang YN, Li J and Gao FG: Interleukin 6 augments lung cancer
chemotherapeutic resistance via ataxia-telangiectasia
mutated/NF-kappaB pathway activation. Cancer Sci. 105:1220–1227.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hsia SM, Yu CC, Shih YH, Chen M Yuanchien,
Wang TH, Huang YT and Shieh TM: Isoliquiritigenin as a cause of DNA
damage and inhibitor of ataxia-telangiectasia mutated expression
leading to G2/M phase arrest and apoptosis in oral squamous cell
carcinoma. Head Neck. 38:(Suppl 1). E360–E371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang H, Zhang X, Teng L and Legerski RJ:
DNA damage checkpoint recovery and cancer development. Exp Cell
Res. 334:350–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stark GR and Taylor WR: Control of the
G2/M transition. Mol Biotechnol. 32:227–248. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oak CH, Wilson D, Lee HJ, Lim HJ and Park
EK: Potential molecular approaches for the early diagnosis of lung
cancer (review). Mol Med Rep. 6:931–936. 2012.PubMed/NCBI
|
|
17
|
Tian W and Chen SY: Recent advances in the
molecular basis of anti-neoplastic mechanisms of oridonin. Chin J
Integr Med. 19:315–320. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li D, Xu S, Cai H, Pei L, Zhang H, Wang L,
Yao H, Wu X, Jiang J, Sun Y and Xu J: Enmein-type diterpenoid
analogs from natural kaurene-type oridonin: Synthesis and their
antitumor biological evaluation. Eur J Med Chem. 64:215–221. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ikezoe T, Chen SS, Tong XJ, Heber D,
Taguchi H and Koeffler HP: Oridonin induces growth inhibition and
apoptosis of a variety of human cancer cells. Int J Oncol.
23:1187–1193. 2003.PubMed/NCBI
|
|
20
|
Chen G, Wang K, Yang BY, Tang B, Chen JX
and Hua ZC: Synergistic antitumor activity of oridonin and arsenic
trioxide on hepatocellular carcinoma cells. Int J Oncol.
40:139–147. 2012.PubMed/NCBI
|
|
21
|
Liu Y, Liu YZ, Zhang RX, Wang X, Meng ZJ,
Huang J, Wu K, Luo JY, Zuo GW, Chen L, et al: Oridonin inhibits the
proliferation of human osteosarcoma cells by suppressing
Wnt/β-catenin signaling. Int J Oncol. 45:795–803. 2014.PubMed/NCBI
|
|
22
|
Gao FH, Liu F, Wei W, Liu LB, Xu MH, Guo
ZY, Li W, Jiang B and Wu YL: Oridonin induces apoptosis and
senescence by increasing hydrogen peroxide and glutathione
depletion in colorectal cancer cells. Int J Mol Med. 29:649–655.
2012.PubMed/NCBI
|
|
23
|
Bu HQ, Liu DL, Wei WT, Chen L, Huang H, Li
Y and Cui JH: Oridonin induces apoptosis in SW1990 pancreatic
cancer cells via p53- and caspase-dependent induction of p38 MAPK.
Oncol Rep. 31:975–982. 2014.PubMed/NCBI
|
|
24
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kikuchi K, Hettmer S, Aslam MI, Michalek
JE, Laub W, Wilky BA, Loeb DM, Rubin BP, Wagers AJ and Keller C:
Cell-cycle dependent expression of a translocation-mediated fusion
oncogene mediates checkpoint adaptation in rhabdomyosarcoma. PLoS
Genet. 10:e10041072014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schafer KA: The cell cycle: A review. Vet
Pathol. 35:461–478. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Falck J, Coates J and Jackson SP:
Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of
DNA damage. Nature. 434:605–611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jazayeri A, Falck J, Lukas C, Bartek J,
Smith GC, Lukas J and Jackson SP: ATM- and cell cycle-dependent
regulation of ATR in response to DNA double-strand breaks. Nat Cell
Biol. 8:37–45. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee JH and Paull TT: ATM activation by DNA
double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science.
308:551–554. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oren M and Rotter V: Introduction: p53-the
first twenty years. Cell Mol Life Sci. 55:9–11. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meek DW: Regulation of the p53 response
and its relationship to cancer. Biochem J. 469:325–346. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Simpson ER and Brown KA: p53:
Protection against tumor growth beyond effects on cell cycle and
apoptosis. Cancer Res. 75:5001–5007. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Douglas P, Zhong J, Ye R, Moorhead GB, Xu
X and Lees-Miller SP: Protein phosphatase 6 interacts with the
DNA-dependent protein kinase catalytic subunit and dephosphorylates
gamma-H2AX. Mol Cell Biol. 30:1368–1381. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kinner A, Wu W, Staudt C and Iliakis G:
Gamma-H2AX in recognition and signaling of DNA double-strand breaks
in the context of chromatin. Nucleic Acids Res. 36:5678–5694. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bakkenist CJ and Kastan MB: DNA damage
activates ATM through intermolecular autophosphorylation and dimer
dissociation. Nature. 421:499–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reinhardt HC and Yaffe MB: Kinases that
control the cell cycle in response to DNA damage: Chk1, Chk2 and
MK2. Curr Opin Cell Biol. 21:245–255. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gerić M, Gajski G and Garaj-Vrhovac V:
γ-H2AX as a biomarker for DNA double-strand breaks in
ecotoxicology. Ecotoxicol Environ Saf. 105:13–21. 2014. View Article : Google Scholar : PubMed/NCBI
|