Introduction
Pulmonary arterial hypertension (PAH) is a
progressive and life-threatening disease that results in a
progressive increase in pulmonary vascular resistance, cardiac
failure and mortality (1). Chronic
hypoxia is an important contributing factor to PAH, which is
characterized by pulmonary vascular remodeling (2). The remodeling process includes
proliferation of intima, hypertrophy of the medial and adventitial
layers, and deposition of extracellular matrix (3,4).
Pulmonary arteries display complex structural and functional
changes in PAH, and endothelial cell dysfunction is important in
disease progression; various cell types, growth factors and their
receptors have been implicated in the development of PAH (5,6).
Vascular smooth muscle cells (VSMCs) are present in the medial wall
of blood vessels and are normally quiescent, expressing a
differentiated phenotype to maintain vascular tone under normal
physiological conditions. However, under pathological conditions,
VSMCs can switch to a ‘synthetic’ phenotype in which they secrete
inflammatory cytokines and contribute to the vascular pathogenesis
(7). Unfortunately, few therapies
have, so far, proven to be effective against pulmonary arterial
structure remodeling following the development of PAH.
Galectin-3 (Gal-3) is an important member of the
lectin family, and is composed of a highly conserved N-terminal
domain and a C-terminal carbohydrate recognition domain that
preferentially interacts with β-galactosides (8). It is expressed in various cell types,
including fibroblasts, endothelial cells and inflammatory cells
(9–11), within the cytoplasm, nucleus, and
extracellular space, and binds to the cell surface (12). Gal-3 is involved in numerous
physiological and pathological processes, and has been demonstrated
to be a central contributor to the progression of atherosclerotic
plaques by amplification of key proinflammatory molecules in the
aorta (13). Furthermore, Gal-3 is
closely associated with cardiac dysfunction via induction of
cardiac fibroblast proliferation, collagen deposition and
ventricular dysfunction (14).
However, to the best of our knowledge, its effects on PAH have not
thus far been investigated. Considering the pathophysiology of PAH
and the physiological role of Gal-3, it is reasonable to
hypothesize that Gal-3 is associated with the pathogenesis of PAH,
an angioproliferative vasculopathy.
In the present study, Gal-3 was hypothesized to be
involved in hypoxia-induced PAH. The role and underlying mechanism
of Gal-3 in hypoxia-induced PAH was investigated in vitro
and in vivo.
Materials and methods
Animals
The study was approved by the ethics committee of
the Medical College of Qingdao University (approval no. 2015–107;
Qingdao, China). A total of 45 mice were obtained from Jackson
Laboratory (Bar Harbor, ME, USA): 30 male C57BL/6J mice, aged 10
weeks, 20–25 g; and 15 male Gal-3−/− mice, aged 10
weeks, 20–25 g (15). The C57BL/6J
mice were randomly divided into two groups: The normal control
group (15 mice) and the hypoxia group (15 mice). The C57BL/6J mice
in the control group were exposed to normoxic conditions, whereas
the C57BL/6J mice in the hypoxia group and the 15
Gal-3−/− mice were exposed to hypoxic conditions (10%
O2) using a 500-liter ventilated chamber (Flufrance
Apparatus, Cachan, France) for 4 weeks as described previously
(16). All animals were kept in
the same room on a 12 h light: 12 h dark cycle at 22°C, and had
ad libitum access to standard mouse chow and water. The
investigation was performed in accordance with the Guide for the
Care and Use of Laboratory Animals (publication no. 82–23, revised
in 1996; National Institutes of Health, Bethesda, MD, USA).
Measurement of right ventricular
systolic pressure (RVSP) and Fulton's index
Prior to euthanasia by intraperitoneal
administration of ketamine (100 mg/kg) and xylazine (10 mg/kg), the
RVSP of mice was measured by right heart catheterization. Briefly,
mice were anesthetized with intraperitoneal injection of
pentobarbital sodium (50 mg/kg), then orally intubated and
ventilated using a rodent respirator (Harvard Apparatus, Holliston,
MA, USA). The tidal volume was set at 250 µl, and the respiratory
rate was set at 120 breaths per minute. The right jugular vein was
isolated, and a small polyethylene catheter was passed through a
small transverse cut and advanced into the right ventricle. RVSP
was recorded using a miniature pressure transducer (MPCU-200;
Millar, Inc., Houston, TX, USA) digitized by a data acquisition
system (ML800 Powerlab 16/30, ADInstruments, Ltd., Oxford, UK).
Following measurement of RVSP, the right ventricle (RV), left
ventricle (LV) and the septum (S) were isolated and weighed, and
the Fulton's index was calculated according to the following
formula: Fulton's index = RV/(LV + S).
Cell culture and cell
transfection
Human pulmonary arterial endothelial cells (HPAECs,
catalog no. PCS-100-022) and human pulmonary arterial smooth muscle
cells (HPASMCs, catalog no. PCS-100-023) were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA) and
cultured in endothelial cell medium or smooth muscle cell medium
(ScienCell Research Laboratories, San Diego, CA, USA) containing
10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C in a 5% CO2 and 95% air
atmosphere. Cells were used for the subsequent experiments up to
passage 4. THP-1 monocytes (catalog no. TIB-202; ATCC) were
cultured in RPMI-1640 medium (Gibco; Thermo Fisher Scientific,
Inc.) containing 2 mM L-glutamine, 10% FBS and 100 U/ml penicillin
and 100 µg/ml streptomycin (Gibco; Thermo Fisher Scientific,
Inc.).
To inhibit Gal-3 expression in HPAECs and HPASMCs,
cells were transfected with 50 nM small interfering RNA (siRNA)
negative control (si-NC) or 50 nM Gal-3 siRNA (Shanghai GenePharma,
Shanghai, China) in Optimem medium (Invitrogen; Thermo Fisher
Scientific, Inc.) using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) for 24 h at 37°C. The sequences for siRNA
were as follows: Gal-3 siRNA, 5′-GCUCCAUGAUGCGUUAUCU-3′, and si-NC,
5′-UUGAUGUGUUUAGUCGCUA-3′. HPAECs and HPASMCs (2×106
cells) were then exposed to normoxic conditions (control group) or
hypoxic conditions (10% O2, 5% CO2) in a cell
culture incubator at 37°C for 48 h (14).
Measurement of HPASMC proliferation
and flow cytometric analysis
For cell counting, HPASMCs were seeded in a 6-well
plate at a density of 5,000 cells/well, then exposed to normal or
hypoxic conditions, with or without Gal-3 siRNA transfection. At
the end of the experiment, cells were washed in PBS, harvested with
trypsin, and counted using a hemocytometer. Cell cycle distribution
was detected through flow cytometry using a cell cycle analysis kit
(catalog no. C1052; Beyotime Institute of Biotechnology, Haimen,
China). Briefly, HPASMCs were seeded in a 6-well plate (Corning
Incorporated, Corning, NY, USA) at a density of 5,000 cells/well at
37°C for 24 h and preincubated with 25 µM diindolylmethane for 1 h,
then trypsinized and fixed with 70% ethanol at 4°C, overnight. The
fixed cells were collected by centrifugation at 800 × g for
15 min, washed once in PBS and incubated with 1 ml propidium iodide
(PI) staining buffer (20 µg/ml PI and 50 µg/ml RNase A), then
analyzed with a fluorescence-activated cell sorter (FACS). The cell
cycle distributions were analyzed using Multicycle AV software
version 1.0 (Phoenix Flow Systems, San Diego, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from homogenized pulmonary
arteries using TRIzol reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). Purified RNA (1 µg) was treated with DNase and
reverse transcribed using RevertAid first strand cDNA synthesis kit
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. mRNA expression was analyzed by RT-qPCR with iQ SYBR
Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
qPCR was performed using the following primers: Cyclin D1, forward
5′-CAGACCAGCCTAACAGATTTC-3′, reverse 5′-TGACCCACAGCAGAAGAAG-3′;
p27, forward 5′-CTTGGAGAAGCACTGCCGAGAT-3′, reverse
5′-CCCTGGACACTGCTCCGCTA-3′; Gal-3, forward
5′-GTTATCTGGGTCTGGAAACC-3′, reverse 5′-TCTGTTTGCATTGGGCTTCACC-3′;
and β-actin, forward 5′-ATCATGTTTGAGACCTTCAACA-3′ and reverse
5′-CATCTCTTGCTCGAAGTCCA-3′. Amplification, detection, and data
analysis involved the use of the iCycler version 3.1 real-time PCR
system (Bio-Rad Laboratories, Inc.). PCR amplification cycling
conditions were: 95°C for 5 min, 36 cycles at 95°C for 10 sec,
annealing at 56°C for 30 sec and elongation at 72°C for 30 sec. The
relative expression of genes was obtained using the
2−ΔΔCq calculation method (17). Each sample was analyzed in
triplicate, and the expression was normalized to that of
β-actin.
Western blot analysis
Total protein was extracted from homogenized murine
pulmonary arteries and HPASMCs using radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology), then
centrifuged at 10,000 × g for 5 min and the supernatant was
collected. The protein concentrations were assayed by bicinchoninic
assay (Beyotime Institute of Biotechnology). The samples (50 µg per
lane) were separated on 10% SDS-polyacrylamide gels and
electrophoretically transferred onto nitrocellulose membranes (EMD
Millipore, Billerica, MA, USA). Membranes were blocked with 5%
non-fat milk for 2 h at room temperature, then washed three times
in TBS-0.1% Tween 20 for 10 min, and incubated with the appropriate
primary antibodies: Rabbit anti-Gal-3 (catalog no. 87985; 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), rabbit
anti-intercellular adhesion molecule 1 (ICAM-1; catalog no. 4915;
1:1,000; Cell Signaling Technology, Inc.), rabbit anti-cyclin D1
(catalog no. 2978; 1:1,000; Cell Signaling Technology, Inc.),
rabbit anti-p27 (catalog no. ab32034; 1:500; Abcam, Cambridge MA,
USA), rabbit anti-α-smooth muscle actin (SMA; catalog no. 14968;
1:1,000; Cell Signaling Technology, Inc.), rabbit anti-smooth
muscle calponin (CNN; catalog no. ab46794; 1:1,000; Abcam) or
rabbit anti-β-actin (catalog no. 4970; 1:1,000; Cell Signaling
Technology, Inc.) at 4°C overnight. Membranes were then washed
three times in TBS+0.1% Tween 20, then incubated with a horseradish
peroxidase-conjugated secondary antibodies (catalog nos. ZB-2301
and ZB-2305; 1:1,000, ZSGB-BIO, Beijing, China) for 2 h at 25°C.
Immune complexes were detected using enhanced chemiluminescence
(EMD Millipore) and analyzed by Image-Pro Plus 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA).
ELISA
Cell culture supernatants were obtained by
centrifugation at 1,000 × g for 10 min at 4°C. Secretion of
tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)) was
determined using ELISA kits according to the manufacturer's
protocols (catalog no. #070133, Uscn Life Sciences, Inc., Wuhan,
China and catalog no. FHK0016, Jiamay Biotech Co. Ltd., Beijing,
China, respectively). All operations were performed at room
temperature. Mean absorbance for standards and samples was assessed
in duplicate. The color reaction was detected using a Varioskan
Flash multifunction plate reader (Thermo Fisher Scientific,
Inc.).
Monocyte adhesion assay
THP-1 monocyte adhesion assays were performed as
previously described (18).
Briefly, THP-1 cells (5×105 cells/ml) were labeled with
10 µM 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein,
acetoxymethyl ester fluorescent dye (Beyotime Institute of
Biotechnology) in serum-free RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) for 45 min at 37°C with frequent agitation.
Following exposure to hypoxia in the presence or absence of siRNA,
the HPAEC monolayers (90% confluence) were washed with endothelial
cell medium and the THP-1 cells (5×105 cells) were
added. Following incubation for 45 min at 37°C, unbound monocytes
were removed by washing with PBS, cells were then fixed with 4%
paraformaldehyde and mounted onto a glass coverslip. Bound
monocytes were quantified by counting the cells under a fluorescent
microscope (two wells for each condition, with five fields of view
assessed).
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three replicates. Analyses were performed using SPSS
version 13.0 for Windows (SPSS, Inc., Chicago, IL, USA). All
statistical comparisons were performed using one-way analysis of
variance with least significant difference post-hoc analysis or
Chi-squared tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Gal-3 inhibition reduces the
hypoxia-induced increase in RVSP and Fulton's index in vivo
Gal-3 mRNA and protein expression levels were
examined in three groups of mice: i) Wild type mice in normoxic
conditions; ii) wild type mice in hypoxic conditions; and iii)
Gal-3−/− mice in hypoxic conditions. Gal-3 mRNA and
protein expression was undetectable in Gal-3−/− mice
(Fig. 1A and B), whereas Gal-3
mRNA and protein expression in hypoxic wild type mice were
significantly increased compared with the normoxic control group
(P<0.001 and P<0.001, respectively; Fig. 1A and B). The hypothesis that Gal-3
deletion may alleviate PAH in mice was then examined through
measurement of hemodynamic parameters prior to euthanasia. No
significant difference was observed in mean blood pressure and
heart rate between the groups (data not shown). Compared with
normoxic wild type control mice, mice in the wild type hypoxia
group had a higher RVSP (P<0.001; Fig. 1C). However, the RVSP was
significantly reduced in hypoxic Gal-3−/− mice compared
with hypoxic wild type mice (P=0.002; Fig. 1C). It has previously been
demonstrated that exposure to chronic hypoxia promotes right
ventricular hypertrophy (19); in
the present study, Fulton's index was used to assess right
ventricular hypertrophy. Hypoxia was demonstrated to significantly
increase the Fulton's index in wild type mice compared with
normoxic wild type control mice (P<0.001; Fig. 1D), whereas it was reduced in
hypoxic Gal-3−/− mice compared with hypoxic wild type
mice (P<0.001; Fig. 1D). These
findings suggest that deletion of Gal-3 reduces hypoxia-induced
increases in RVSP and RV hypertrophy.
Hypoxia induces in vitro expression of
Gal-3
The effect of hypoxia on Gal-3 expression was then
investigated in vitro. Gal-3 mRNA and protein expression
levels were detected in HPAECs and HPASMCs cultured under normoxic
and hypoxic conditions. HPAECs and HPASMCs were also transfected
with si-Gal-3 siRNA to inhibit Gal-3 expression and si-NC, and
cultured under hypoxic conditions. Compared with the normoxic
control group, hypoxia significantly increased Gal-3 mRNA and
protein expression levels in untransfected cells (P<0.05;
Fig. 2), whereas hypoxic cells
transfected with si-Gal-3 demonstrated significantly reduced levels
of Gal-3 mRNA and protein expression compared with untransfected
hypoxic cells (P<0.05; Fig.
2).
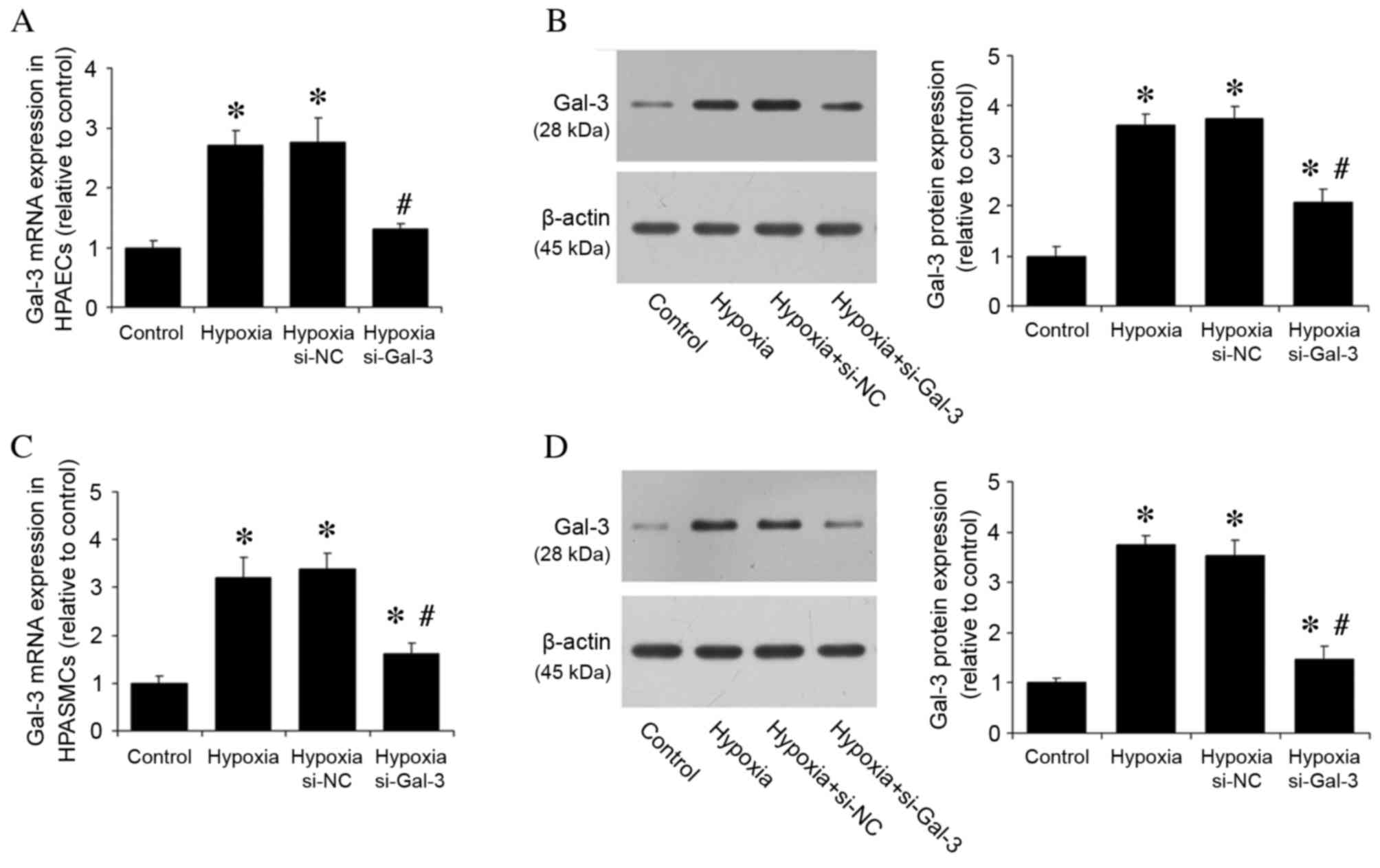 | Figure 2.Hypoxia increases Gal-3 mRNA and
protein expression in HPAECs and HPASMCs. (A) Gal-3 mRNA expression
in HPAECs was assessed by RT-qPCR, with quantitation relative to
β-actin. (B) Gal-3 protein expression in HPAECs was assessed by
western blot analysis, with quantitation relative to β-actin.
Hypoxia increased mRNA and protein expression levels of Gal-3 in
HPAECs, whereas Gal-3 inhibition by siRNA reduced expression. (C)
Gal-3 mRNA expression in HPASMCs was assessed by RT-qPCR, with
quantitation relative to β-actin. (D) Gal-3 protein expression in
HPASMCs was assessed by western blot analysis, with quantitation
relative to β-actin. Hypoxia increased Gal-3 mRNA and protein
expression levels in HPASMCs, whereas Gal-3 inhibition reduced
expression. Data are presented as the mean ± standard deviation of
three independent replicates. *P<0.05 vs. normoxic control;
#P<0.05 vs. hypoxia. Gal-3, galectin-3; HPAECs, human
pulmonary arterial endothelial cells; HPASMCs, human pulmonary
arterial smooth muscle cells; si-NC, negative control small
interfering RNA; si-Gal-3, Gal-3 small interfering RNA; RT-qPCR,
reverse transcription-quantitative polymerase chain reaction. |
Gal-3 inhibition reduces the
hypoxia-induced inflammatory response
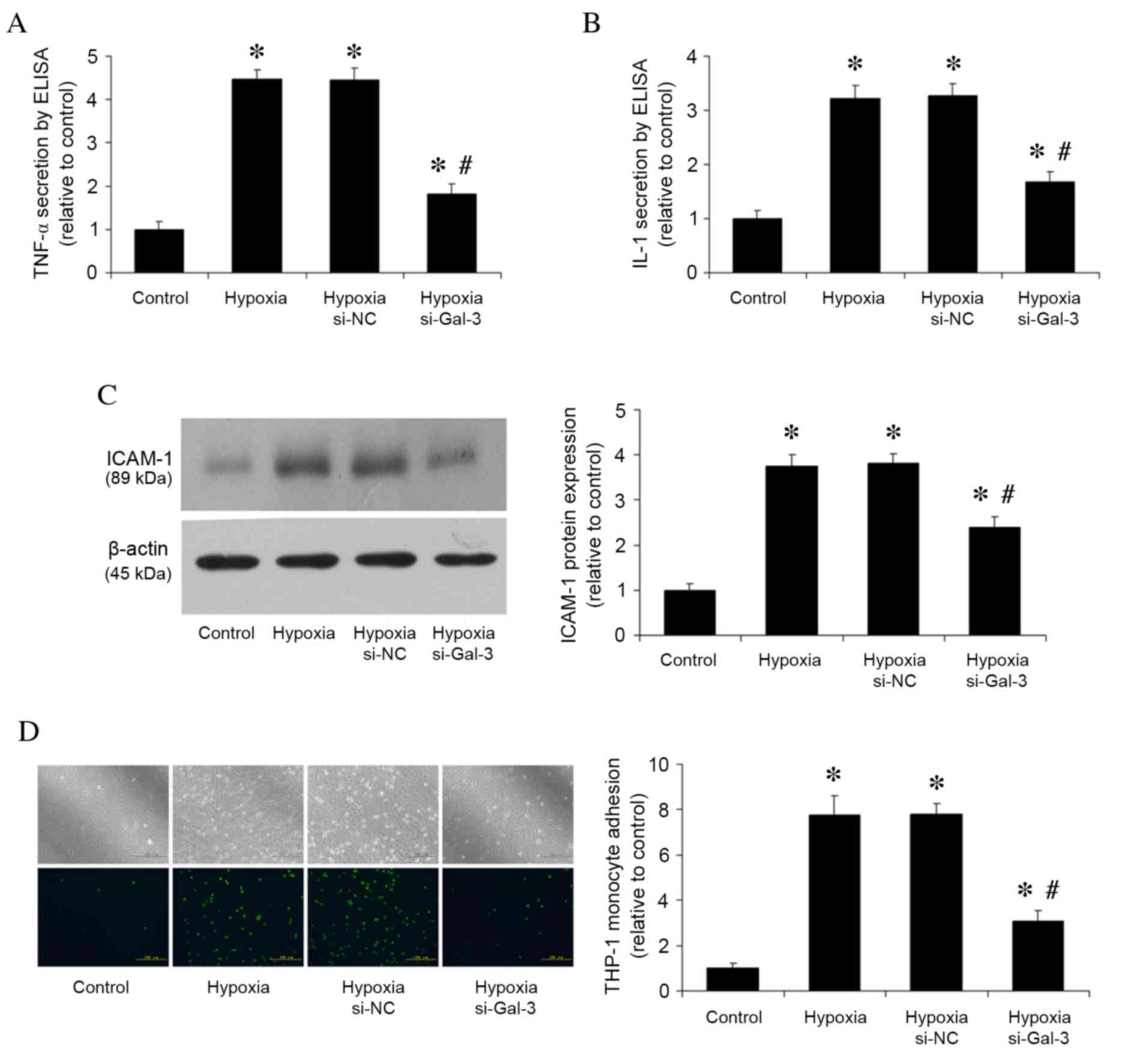
Following 48 h stimulation of HPAECs with hypoxia,
TNF-α and IL-1 secretion levels were assessed by ELISA (Fig. 3A and B), ICAM-1 protein expression
levels were analyzed by western blotting (Fig. 3C), and THP-1 monocyte adhesion was
analyzed by monocyte adhesion assay (Fig. 3D). Compared with the normoxic
control group, hypoxia increased TNF-α and IL-1 secretion
(P<0.001 and P<0.001, respectively; Fig. 3A and B, respectively), ICAM-1
protein expression (P<0.001; Fig.
3C), and the number of adhered THP-1 cells (P<0.001;
Fig. 3D). Gal-3 inhibition in
hypoxic cells resulted in reduced TNF-α and IL-1 secretion
(P<0.001 and P<0.001, respectively; Fig. 3A and B), reduced ICAM-1 protein
expression (P<0.001; Fig. 3C),
and fewer adhered THP-1 cells (P<0.05; Fig. 3D) compared with untransfected
hypoxic cells.
Gal-3 inhibition reduces
hypoxia-induced HPASMC proliferation
Following 48 h of hypoxic stimulation of HPASMCs,
cells were harvested and counted using a hemocytometer (Fig. 4A). The mean number of viable
HPASMCs in the hypoxia group was 1.65-fold higher than that in the
normoxic control group (P=0.002; Fig.
4B). However, Gal-3 inhibition in hypoxic cells significantly
reduced the mean number of viable HPASMCs compared with
untransfected hypoxic cells (P=0.019; Fig. 4B).
Gal-3 inhibition results in HPASMC
arrest in G0/G1-phase under conditions of
hypoxia
Cell proliferation depends on cell cycle transition
from G0/G1-phase to S-phase and
G2/M-phase. The effect of Gal-3 inhibition on the cell
cycle distribution of HPASMCs was, therefore, analyzed. Hypoxia
reduced the number of cells in G0/G1-phase
compared with normoxic control cells (P=0.032; Fig. 4C), whereas Gal-3 inhibition in
hypoxic cells arrested more HPASMCs in the
G0/G1-phase (P=0.046; Fig. 4C).
Gal-3 inhibition reduces cyclin D1
expression, but increases p27 expression
The underlying mechanism of Gal-3 modulation on cell
proliferation and the cell cycle was then investigated. Previous
studies have demonstrated that cyclin D1 and p27 are important in
cell proliferation and the cell cycle (20); therefore, their mRNA and protein
expression levels were examined in vitro. RT-qPCR revealed
that mRNA expression levels of cyclin D1 were significantly reduced
by Gal-3 inhibition in hypoxic cells compared with untransfected
hypoxic cells (P=0.002; Fig. 5A),
whereas Gal-3 inhibition in hypoxic cells increased p27 mRNA
expression compared with untransfected hypoxic cells (P=0.048;
Fig. 5B). Western blot analysis of
protein expression levels revealed the same pattern of reduced
cyclin D1 protein expression and increased p27 protein expression
in hypoxic Gal-3 inhibited cells compared with untransfected
hypoxic cells (P<0.001 and P=0.003, respectively; Fig. 5C and D). These results suggest that
Gal-3 inhibition may alleviate PAH via regulation of cell
cycle.
Gal-3 inhibition suppresses HPASMC
transition from ‘contractile’ to ‘synthetic’ phenotype
Under pathological conditions, VSMCs can switch to a
‘synthetic’ phenotype with a reduced expression of SMA and CNN, in
which they secrete inflammatory cytokines, and which contributes to
vascular pathogenesis (21). To
examine the effects of Gal-3 on phenotype switching, protein
expression levels of SMA and CNN were measured in HPASMCs. Compared
with the control group, hypoxia reduced the expression of SMA and
CNN (P<0.001 and P<0.001, respectively; Fig. 6A and B), whereas Gal-3 inhibition
increased their expression (P=0.017 and P=0.021, respectively;
Fig. 6A and B). These results
suggest a critical function of Gal-3 during the phenotype
transition of HPASMCs.
Discussion
PAH, which is characterized by a persistent increase
in pulmonary artery pressure and pulmonary vascular remodeling, is
a progressive disease that is associated with a poor prognosis
(22). PAH is diagnosed when the
mean pulmonary arterial pressure exceeds 25 mmHg, as measured by
right-heart catheterization (18).
Although advances have been made in the understanding of PAH and
development of treatments, research into effective therapies is
still required to improve the long-term survival of patients with
fewer side effects than current treatments exert. It has been
demonstrated that Gal-3 expression is increased in the left
ventricle in the early ischemic period, which may be part of the
prosurvival gene expression profile transcribed by
hypoxia-inducible factor 1a (23).
Increased expression of Gal-3 protects against cell death under
conditions of hypoxia and nutrient deprivation (24). In the present study, Gal-3 was
demonstrated to be associated with hypoxia-induced PAH. Exposure to
chronic hypoxia resulted in significantly elevated RVSP and
increased Fulton's index in wild type mice. However, the increase
was inhibited in Gal-3−/− mice. Furthermore, Gal-3
inhibition reduced the hypoxia-induced inflammatory response in
HPAECs and reduced HPASMC cell proliferation by arresting cells in
G0/G1-phase. Gal-3 inhibition also resulted
in reduced expression of cyclin D1 and increased p27 expression in
HPASMCs, and inhibited the switch of HPASMCs to a ‘synthetic’
phenotype. These novel findings contribute to the understanding of
the mechanism by which Gal-3 inhibition ameliorates hypoxia-induced
PAH.
Inflammation is important during PAH disease
progression (25). There is an
increase in serum levels of several chemokines and cytokines
related to inflammatory processes during PAH (26). Cytokines are a large group of
signaling proteins, which regulate numerous biological processes,
including inflammation, immunity and hematopoiesis. Cytokines, such
as TNF-α, IL-8 and monocyte chemotactic protein-1, contribute to
leukocyte recruitment, endothelin-1 induction, and smooth muscle
cell (SMC) proliferation (26,27).
In a previous study, rats treated with a TNF-α inhibitor
demonstrated amelioration in pulmonary hemodynamics, right
ventricular hypertrophy and pulmonary inflammation (28). In the present study, hypoxia was
demonstrated to increase TNF-α and IL-1 secretion, increase ICAM-1
protein expression and increase adhesion of THP-1 monocytes in
vitro, whereas Gal-3 inhibition by siRNA was demonstrated to
reduce the inflammatory response. This may be one mechanism by
which Gal-3 inhibition protects against PAH.
Similar to other vasculopathies, PAH is
characterized by severe angioproliferative vascular remodeling
(29). The proliferation of
HPASMCs is important in the progression of PAH; effective
inhibition of aberrant HPASMC proliferation can delay, and even
halt, the deteriorative progress of PAH (5). In the present study, under conditions
of hypoxia, few HPASMCs remained in G0/G1
phase and more cells entered the mitotic cycle, whereas Gal-3
inhibition reduced hypoxia-induced HPASMC proliferation, partially
through cell cycle arrest in G0/G1-phase. The
possible mechanism underlying this effect was then investigated.
The cell cycle is regulated by cyclin-dependent kinases (CDK) and
CDK inhibitors, which have been key therapeutic targets in treating
vascular proliferation-associated diseases (16). Cyclin D1 is the key cell cycle
control gene that facilitates the transition of cells from the
G1 phase into the S phase (18). A previous study demonstrated that
inhibition of cyclin D1 could inhibit VSMC proliferation (30). The present study demonstrated that
hypoxia increased the mRNA and protein expression of cyclin D1,
whereas Gal-3 inhibition significantly reduced cyclin D1
expression. p27, which is an important CDK inhibitor, can
effectively inhibit cyclin D1 activity and negatively regulate
G1 progression in cells (31). It has been demonstrated that p27 is
one of the potent inhibitors of VSMC growth in vivo and
in vitro (32,33). Fouty et al (29) demonstrated that p27 modulates PASMC
proliferation during mitogenic stimulation, and that overexpression
of p27 decreases PASMC proliferation. The present study
demonstrated that Gal-3 inhibition increases p27 mRNA and protein
expression.
The principal phenotype of SMCs is contractile,
which preserves vasodilation and blood flow regulation under normal
physiological conditions. However, in pathological conditions, SMCs
can transform from the differentiated contractile phenotype to a
synthetic state, which is characterized by high proliferation,
migration and extracellular matrix production. During this process,
the contractile ability of SMCs is reduced, resulting in a lack of
resistance to environmental stimulation (22). Normally, VSMCs express the
contractile phenotype by regulating specific genes, including SMA,
CNN and SM22α (21). As a response
to stimuli including inflammation, oxidative stress and shear
stress, quiescent contractile cells reduce the expression of
SMC-specific genes to promote their proliferation, migration and
collagen synthesis, in order to remodel the phenotypic state into
the synthetic state (34,35). However, aberrant phenotype
transitioning leads to pulmonary arterial remodeling. In the
present study, hypoxia was observed to reduce SMA and CNN protein
expression, whereas Gal-3 inhibition increased their expression.
These results suggested that Gal-3 inhibition could suppress the
transformation of HPASMCs from a contractile to a synthetic
phenotype.
In conclusion, the present study presents evidence
that Gal-3 inhibition reduces the increased RVSP and alleviated RV
hypertrophy of mice with PAH. In vitro Gal-3 inhibition was
also demonstrated to reduce the hypoxia-induced inflammatory
response and reduce HPASMC proliferation by decreasing cyclin D1
expression and increasing p27 expression. Gal-3 inhibition also
maintained a contractile phenotype in HPASMCs. These findings may
lead to a useful therapeutic intervention for the treatment of
pulmonary hypertensive disorders.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81400340).
References
|
1
|
Humbert M, Morrell NW, Archer SL, Stenmark
KR, MacLean MR, Lang IM, Christman BW, Weir EK, Eickelberg O,
Voelkel NF and Rabinovitch M: Cellular and molecular pathobiology
of pulmonary arterial hypertension. J Am Coll Cardiol. 43:(12 Suppl
S). 13S–24S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sudar E, Dobutovic B, Soskic S, Mandusic
V, Zakula Z, Misirkic M, Vucicevic L, Janjetovic K, Trajkovic V,
Mikhailidis DP and Isenovic ER: Regulation of inducible nitric
oxide synthase activity/expression in rat hearts from
ghrelin-treated rats. J Physiol Biochem. 67:195–204. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Orlandi A, Bochaton-Piallat ML, Gabbiani G
and Spagnoli LG: Aging, smooth muscle cells and vascular
pathobiology: Implications for atherosclerosis. Atherosclerosis.
188:221–230. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stenmark KR, Fagan KA and Frid MG:
Hypoxia-induced pulmonary vascular remodeling: Cellular and
molecular mechanisms. Circ Res. 99:675–691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Luo Y, Xu DQ, Dong HY, Zhang B, Liu Y, Niu
W, Dong MQ and Li ZC: Tanshinone IIA inhibits hypoxia-induced
pulmonary artery smooth muscle cell proliferation via
Akt/Skp2/p27-associated pathway. PLoS One. 8:e567742013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasa M, Fichtlscherer S, Adler K, Aicher
A, Martin H, Zeiher AM and Dimmeler S: Increase in circulating
endothelial progenitor cells by statin therapy in patients with
stable coronary artery disease. Circulation. 103:2885–2890. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen B, Calvert AE, Cui H and Nelin LD:
Hypoxia promotes human pulmonary artery smooth muscle cell
proliferation through induction of arginase. Am J Physiol Lung Cell
Mol Physiol. 297:L1151–L1159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dennis JW, Pawling J, Cheung P, Partridge
E and Demetriou M: UDP-N-acetylglucosamine: Alpha-6-D-mannoside
beta1, 6 N-acetylglucosaminyltransferase V (Mgat5) deficient mice.
Biochim Biophys Acta. 1573:414–422. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma UC, Pokharel S, van Brakel TJ, van
Berlo JH, Cleutjens JP, Schroen B, André S, Crijns HJ, Gabius HJ,
Maessen J and Pinto YM: Galectin-3 marks activated macrophages in
failure-prone hypertrophied hearts and contributes to cardiac
dysfunction. Circulation. 110:3121–3128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thijssen VL, Hulsmans S and Griffioen AW:
The galectin profile of the endothelium: Altered expression and
localization in activated and tumor endothelial cells. Am J Pathol.
172:545–553. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wan SY, Zhang TF and Ding Y: Galectin-3
enhances proliferation and angiogenesis of endothelial cells
differentiated from bone marrow mesenchymal stem cells. Transplant
Proc. 43:3933–3938. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dumic J, Dabelic S and Flögel M:
Galectin-3: An open-ended story. Biochim Biophys Acta.
1760:616–635. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Papaspyridonos M, McNeill E, de Bono JP,
Smith A, Burnand KG, Channon KM and Greaves DR: Galectin-3 is an
amplifier of inflammation in atherosclerotic plaque progression
through macrophage activation and monocyte chemoattraction.
Arterioscler Thromb Vasc Biol. 28:433–440. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu X, Murphy TC, Nanes MS and Hart CM:
PPAR{gamma} regulates hypoxia-induced Nox4 expression in human
pulmonary artery smooth muscle cells through NF-{kappa}B. Am J
Physiol Lung Cell Mol Physiol. 299:L559–L566. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Henderson NC, Mackinnon AC, Farnworth SL,
Kipari T, Haslett C, Iredale JP, Liu FT, Hughes J and Sethi T:
Galectin-3 expression and secretion links macrophages to the
promotion of renal fibrosis. Am J Pathol. 172:288–298. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu L, Quinn DA, Garg HG and Hales CA: Gene
expression of cyclin-dependent kinase inhibitors and effect of
heparin on their expression in mice with hypoxia-induced pulmonary
hypertension. Biochem Biophys Res Commun. 345:1565–1572. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dong Y, Sui L, Sugimoto K, Tai Y and
Tokuda M: Cyclin D1-CDK4 complex, a possible critical factor for
cell proliferation and prognosis in laryngeal squamous cell
carcinomas. Int J Cancer. 95:209–215. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Campen MJ, Paffett ML, Colombo ES, Lucas
SN, Anderson T, Nysus M, Norenberg JP, Gershman B, Hesterman J,
Hoppin J and Willis M: Muscle RING finger-1 promotes a maladaptive
phenotype in chronic hypoxia-induced right ventricular remodeling.
PLoS One. 9:e970842014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee JW, Kim HS, Kim S, Hwang J, Kim YH,
Lim GY, Sohn WJ, Yoon SR, Kim JY, Park TS, et al: DACH1 regulates
cell cycle progression of myeloid cells through the control of
cyclin D, Cdk 4/6 and p21Cip1. Biochem Biophys Res Commun.
420:91–95. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hutcheson R, Terry R, Chaplin J, Smith E,
Musiyenko A, Russell JC, Lincoln T and Rocic P: MicroRNA-145
restores contractile vascular smooth muscle phenotype and coronary
collateral growth in the metabolic syndrome. Arterioscler Thromb
Vasc Biol. 33:727–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rensen SS, Doevendans PA and van Eys GJ:
Regulation and characteristics of vascular smooth muscle cell
phenotypic diversity. Neth Heart J. 15:100–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashmi S and Al-Salam S: Galectin-3 is
expressed in the myocardium very early post-myocardial infarction.
Cardiovasc Pathol. 24:213–223. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ikemori RY, Machado CM, Furuzawa KM,
Nonogaki S, Osinaga E, Umezawa K, de Carvalho MA, Verinaud L and
Chammas R: Galectin-3 up-regulation in hypoxic and nutrient
deprived microenvironments promotes cell survival. PLoS One.
9:e1115922014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mor A, Planer D, Luboshits G, Afek A,
Metzger S, Chajec-Shaul T, Keren G and George J: Role of naturally
occurring CD4+ CD25+ regulatory T cells in experimental
atherosclerosis. Arterioscler Thromb Vasc Biol. 27:893–900. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hassoun PM, Mouthon L, Barberà JA,
Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML,
Michelakis ED, Morrell NW, et al: Inflammation, growth factors, and
pulmonary vascular remodeling. J Am Coll Cardiol. 54:(Suppl 1).
S10–S19. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chaouat A, Savale L, Chouaid C, Tu L,
Sztrymf B, Canuet M, Maitre B, Housset B, Brandt C, Le Corvoisier
P, et al: Role for interleukin-6 in COPD-related pulmonary
hypertension. Chest. 136:678–687. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Q, Zuo XR, Wang YY, Xie WP, Wang H
and Zhang M: Monocrotaline-induced pulmonary arterial hypertension
is attenuated by TNF-α antagonists via the suppression of TNF-α
expression and NF-κB pathway in rats. Vascul Pharmacol. 58:71–77.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fouty BW, Grimison B, Fagan KA, Le Cras
TD, Harral JW, Hoedt-Miller M, Sclafani RA and Rodman DM: p27(Kip1)
is important in modulating pulmonary artery smooth muscle cell
proliferation. Am J Respir Cell Mol Biol. 25:652–658. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao Y, Lv M, Lin H, Cui Y, Wei X, Qin Y,
Kohama K and Gao Y: Rho-associated protein kinase isoforms
stimulate proliferation of vascular smooth muscle cells through ERK
and induction of cyclin D1 and PCNA. Biochem Biophys Res Commun.
432:488–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arteaga CL: Cdk inhibitor p27Kip1 and
hormone dependence in breast cancer. Clin Cancer Res. 10:(Suppl).
368S–371S. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Akyurek LM, Boehm M, Olive M, Zhou AX, San
H and Nabel EG: Deficiency of cyclin-dependent kinase inhibitors
p21Cip1 and p27Kip1 accelerates atherogenesis in apolipoprotein
E-deficient mice. Biochem Biophys Res Commun. 396:359–363. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tanner FC, Boehm M, Akyurek LM, San H,
Yang ZY, Tashiro J, Nabel GJ and Nabel EG: Differential effects of
the cyclin-dependent kinase inhibitors p27(Kip1), p21(Cip1) and
p16(Ink4) on vascular smooth muscle cell proliferation.
Circulation. 101:2022–2025. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wali MA and Eid RA: Smooth muscle changes
in varicose veins: An ultrastructural study. J Smooth Muscle Res.
37:123–135. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Naoum JJ, Hunter GC, Woodside KJ and Chen
C: Current advances in the pathogenesis of varicose veins. J Surg
Res. 141:311–316. 2007. View Article : Google Scholar : PubMed/NCBI
|