Introduction
The endocrine disease, congenital adrenal
hyperplasia (CAH), defines a group of syndromes resulting from
inherited defects in one of the five enzymes involved in the
biosynthesis of cortisol from cholesterol. These enzymes include
21α-hydroxylase, 11β-hydroxylase and 17α-hydroxylase/17,20-lyase,
which are associated with P450C21, P45011B, and P45017A1 forms of
cytochrome P450, as well as cholesterol 20,22 desmolase and
3β-hydroxysteroid dehydrogenase (1). As a result of this enzymatic defect,
cortisol levels decrease and the negative feedback control of
adrenocorticotropic hormone (ACTH) levels is absent, which leads to
excess ACTH production to normalize cortisol levels (2). This results in overproduction and
accumulation of steroid precursors and high ACTH levels that lead
to adrenal hyperplasia. The clinical manifestation of the disorder
depends on the levels of these intermediates in the steroid
biosynthetic pathway for the blockade of any one of the steps
(2). CAH should be considered in
infants, children or adolescents in the clinic when evaluating
ambiguous genitalia, sexual infantilism, hypogonadism or
hypertension, particularly when associated with disturbed fluid,
electrolyte and hydrogen homeostasis (3). The most common form of CAH involves
21-hydroxylase deficiency, which accounts for 90–95% of CAH cases,
whilst 17α-hydroxylase/17,20-lyase deficiency is the least common
form of the disorder, <1% of all CAH cases (4).
17α-hydroxylase/17,20-lyase deficiency is caused by
mutations in the cytochrome P450 family 17 subfamily A member 1
(CYP17A1) gene, and is associated with the impaired
production of cortisol and sex steroids (5). The deficiency leads to elevation of
plasma ACTH levels and accumulation of mineralocorticoids, thus
resulting in hypertension, hypokalemia and bilateral adrenal
hyperplasia (5). In addition, the
impaired production of sex steroids leads to ambiguous or female
external genitalia in affected male individuals (46, XY), and
normal genitalia in affected females (46, XX) at birth, but no
sexual development at the expected time of puberty (6,7).
The current study presents a case of a female
Chinese patient (age, 17 years) with a deficiency in
17α-hydroxylase/17,20-lyase caused by a compound heterozygous
mutation (c.985–987 in exon 6, c.1459–1467 in exon 8) in the
CYP17A1 gene.
Case report
A Chinese woman (age, 17 years) was transferred from
Shishou Hospital to the Department of Endocrinology at Tongji
Hospital (Wuhan, China) in June 2015, and presented with primary
amenorrhea and evidence of sexual infantilism. The patient's
parents noticed a lack of breast development, as well as an absence
of axillary and pubic hair since the beginning of puberty. The
patient's parents were non-consanguineous and reported no history
of hypertension or recurrent episodes of periodic paralysis. There
was no reported history of surgery or additional diseases in the
patient. In addition, the patient's 10-year-old sister appeared
normal as she presented normal female sex characteristics, no
hypertension and no periodic paralysis.
Upon physical examination, the patient was tall and
slender, with a height of 163 cm, a weight of 45 kg and a body mass
index of 16.9 kg/m2. Blood pressure was normal (128/99
mmHg), and the patient's skin color was slightly darker when
compared with that of her parents. The patient's breast development
had only progressed to Tanner stage B1P1 and external genitalia
were phenotypically female, but were infantile. A wide palpebral
fissure and a slightly thick upper lip was evident, however there
was no webbed neck or cubitus valgus. Cardiovascular system
examination revealed normal heart sounds and muscular tension test
exhibited grade 5 strength.
Routine laboratory tests revealed a normal white
blood cell count, hemoglobin, platelet count, coagulation function,
urine (specific gravity, PH, protein, glucose and microscopic
analysis) and stools. Blood biochemistry analysis revealed results
within the reference ranges for liver function, renal function,
glucose, lipids and thyroid profiles, with low serum potassium
(3.09 mmol/l, normal range, 3.50–5.10 mmol/l) and mild alkalosis
(pH 7.39, normal range, 7.350–7.450; bicarbonate 26 mmol/l, normal
range, 21.3–24.8 mmol/l). Pelvic ultrasonography revealed a
primordial uterus (1.9×0.5 cm) and hypoplastic ovaries (left,
1.2×0.6 cm, right, 1.6×0.7 cm). Magnetic resonance imaging
demonstrated that the pituitary gland was normal, and bilateral
integration of the adrenal glands was enlarged, as determined by
enhanced computed tomography. Ambulatory blood pressure monitoring
demonstrated that blood pressure was in the normal range (average,
127/85 mmHg). However, the patient's bone age was delayed (age, 12
years). Chromosomal analysis revealed a normal 46, XX female
karyotype.
The patient received endocrine tests, and the
results of blood analysis for adrenal, gonadal and pituitary
hormones, as well as the renin-angiotensin-aldosterone system are
presented in Table I. The patient
presented with low serum cortisol levels and high levels of ACTH,
which were consistent with the results upon re-examination after 1
week. Follicle-stimulating hormone (FSH) and luteinizing hormone
(LH) levels were elevated, but estradiol and testosterone were low,
suggesting a diagnosis of primary gonadal failure
(hypergonadotrophic hypogonadism). In addition, the patient
presented with low 17α-hydroxyprogesterone levels (the precursor of
cortisol), and low levels of adrenal androgens
[dehydroepiandrosterone-sulfate (DHEA-S)] and androstenedione
(Table I). The
renin-angiotensin-aldosterone system test demonstrated that the
patient exhibited normal aldosterone levels and undetectable plasma
renin activity. Blood profiles of the patient's parents and sister
demonstrated that they all exhibited normal 17α-hydroxyprogesterone
levels, and the sister had a normal 46, XX karyotype.
 | Table I.Plasma steroid and pituitary hormone
levels in the patient, and 17α-hydroxyprogesterone levels in the
patient's family members at clinical presentation. |
Table I.
Plasma steroid and pituitary hormone
levels in the patient, and 17α-hydroxyprogesterone levels in the
patient's family members at clinical presentation.
| Plasma
steroid/pituitary hormone | Result | Reference range |
|---|
| FSH (mIU/ml) | 104.41 | Follicular,
3.85–8.78; ovulatory, 4.54–22.51; luteal, 1.79–5.12 |
| LH (mIU/ml) | 26.09 | Follicular,
2.12–10.89; ovulatory, 19.18–103.03; luteal, 1.20–12.86 |
| Estradiol
(pg/ml) | <20 | Follicular, 24–114;
ovulatory, 62–534; luteal, 80℃273 |
| Progesterone
(ng/ml) | 9.89 | Follicular,
0.31–1.52; luteal, 5.16–18.56 |
| Testosterone
(ng/ml) | <0.1 | 0.10–0.75 |
| Prolactin
(ng/ml) | 12.32 | 3.34–26.72 |
| SHBG (nmol/l) | 104 | 18.2–135.5 |
| GH (ng/ml) | 0.354 | 0℃10 |
| IGF-1 (ng/ml) | 236 | 412±80 |
| DHEA-S (nmol/l) | <1 | 3540±1310 |
| Androstenedione
(nmol/l) | <0.01 | Follicular, 2.7±1;
luteal, 5.2±1.5 |
|
17α-hydroxyprogesterone (nmol/l) | 1.07 | Follicular, 1.3±0.25;
luteal, 7.4±2 |
| ACTH (pmol/l) | 15.94 | 1.6–13.9 |
| Cortisol (µg/l) 8:00
a.m. | 35.44 | 62℃194 |
| Cortisol (µg/l) 4:00
p.m. | 34.83 | 23℃123 |
| Renin concentration
(ng/ml/h) | <0 | Recumbent, 0.05–0.79;
upright, 0.93–6.56 |
| Aldosterone
(ng/dl) | 16.5 | Recumbent, 5.9–17.4;
upright, 6.5–29.6 |
| Father:
17α-hydroxyprogesterone (nmol/l) | 8.69 | 3.5±1.2 |
| Mother:
17α-hydroxyprogesterone (nmol/l) mother | 1.88 | Follicular, 1.3±0.25;
luteal, 7.4±2 |
| Sister:
17α-hydroxyprogesterone (nmol/l) | 2.21 | Follicular, 1.3±0.25;
luteal, 7.4±2 |
The clinical manifestations, imaging and laboratory
results appeared to be consistent with a diagnosis of CAH in the
patient, due to the observed 17α-hydroxylase deficiency. In order
to confirm this diagnosis, genetic analysis of the CYP17A1
sequence was performed on the patient and her family members.
Genomic DNAs were extracted from the peripheral
blood leukocytes using the QIAamp DNA Blood Mini kit (Qiagen,
Hilden, Germany) per the manufacturer's protocol. DNA was diluted
to a final concentration of 10 ng/µl for use. All eight exons of
CYP17A1 gene were amplified by polymerase chain reaction
(PCR) using eight pairs of primers (Table II), which were designed with
Primer Premier 5.0 software. The PCR was performed in a volume of
50 µl. Amplifications were sequenced with the BigDye Terminator
v3.1 Cycle Sequence kit (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham MA, USA) using an ABI3130xl Genetic
Analyzer (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Finally, the Chromas Lite 2.01 software was used to identify
mutation candidates, which was confirmed by two independent
observers.
| Table II.Primers for CYP17A1 amplification. |
Table II.
Primers for CYP17A1 amplification.
| Exon | Sequence | Product size
(bp) |
|---|
| 1 | F 5′
CTTGTGCCCTAGAGTGCCA 3′ |
|
|
| R 5′
GAAGGGGGCAGGGAGGAG 3′ | 401 |
| 2 | F 5′
GAAGGAAAGCAGGGACCAGA 3′ |
|
|
| R 5′
GGCAGCAGTAGCCAAGAAAA 3′ | 350 |
| 3 | F 5′
CATCTGCTATCTGTCCCCCG 3′ |
|
|
| R 5′
GGCTGGAGCAGGGAAGTAAA 3′ | 419 |
| 4 | F 5′
GCCCTTTGTCCTTTCCCTCA 3′ |
|
|
| R 5′
GGGAACGAAAGGGGTGCTAA 3′ | 468 |
| 5 | F
5′AGTCAGGGACAGAAGTATGGCAG 3′ |
|
|
| R 5′
TGCACAGAAAGCCTGAGAGAATT 3′ | 389 |
| 6 | F 5′
GGAAGGGACTGGACAGGCTC 3′ |
|
|
| R 5′
TGAATGCATCATGGGGCTAGA 3′ | 324 |
| 7 | F 5′
AAGGGCATTTTCCTCACGG 3′ |
|
|
| F 5′
TTGGCAGAGGTGAAGGGGTA 3′ | 291 |
| 8 | F 5′
CTCAACCAGGGCAGAACCAT 3′ |
|
|
| R 5′
GGTGGGGGGTTGTATCTCTAAA 3′ | 429 |
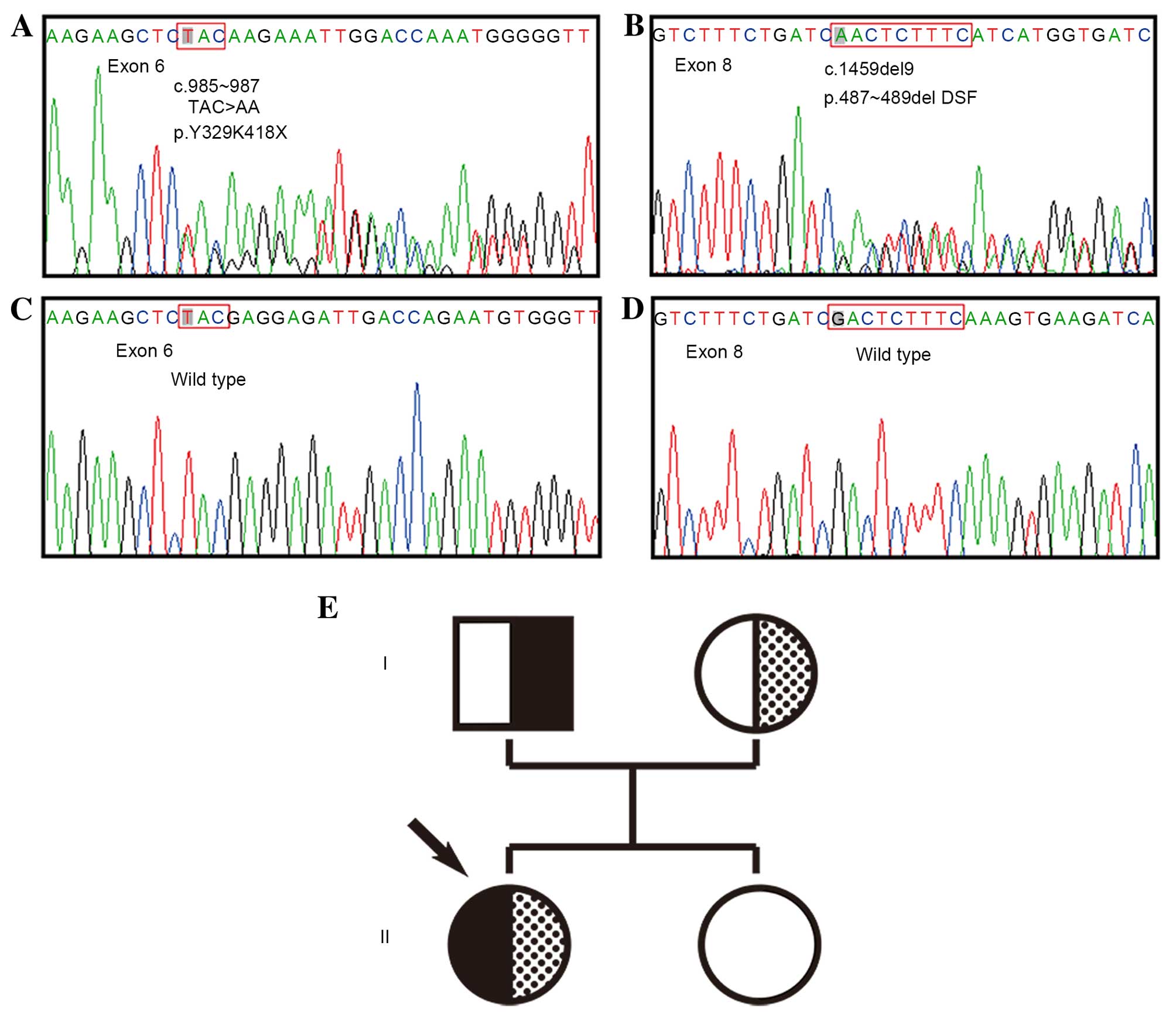
Direct sequencing of PCR products revealed that the
patient harbored the following two compound heterozygous mutations
in the CYP17A1 gene: c.985–987 (TAC>AA) in exon 6,
leading to amino acid alterations Y329K418X, a premature
termination codon; and c.1459–1467 (GACTCTTTC deletion) in exon 8,
leading to deletion of D487-S488-F489 amino acids. Analysis of DNA
from the patient's parents and sister, demonstrated that the
TAC>AA mutation was present in the father, while the 9 bp
deletion was present in the mother. Fortunately, the patient's
sister was homozygous wild-type alleles from her parents (Fig. 1).
Oral prednisolone treatment was started at 5 mg/day,
and estrogen replacement was provided to induce female secondary
sexual development with oral estradiol (Progynova, 1 mg daily). One
year after the diagnosis, the patient's breast development
progressed to Tanner stage B2P2, and she was still amenorrhea and
had infantile external genitalia. The treatment was maintained.
The written informed consent was obtained from the
patient and her family for the use of their samples in the current
study.
Discussion
17α-hydroxylase deficiency is a rare form of CAH,
with an estimated incidence of 1 in 50,000–100,000 individuals and
represents ~1% of all CAH cases (8,9). CAH
is an autosomal recessive disorder that results in decreased
production of cortisol, androgens and estrogens, with a subsequent
increase in ACTH and gonadotropin levels. The majority of patients
with CAH typically present with hypertension and primary gonadal
failure during adolescence and adulthood. However, a few
individuals are reported to be normotensive at the time of
diagnosis (10,11). 17α-hydroxylase deficiency was first
described in a 35-year-old patient in 1966 by Biglieri et al
(12), who was phenotypically
female and presented with hypertension, sexual infantilism and
primary amenorrhea. There have been >200 cases reported in the
literature.
The human CYP17A1 gene is located on
chromosome 10q24.3, and encodes the P450c17 enzyme. The gene spans
6.6 kb, consists of eight exons and has a coding region of 1.6 kb
(13,14). The P450c17 enzyme catalyzes steroid
17α-hydroxylase and 17,20-lyase activities in the adrenal glands
and gonads. Individuals with CAH that present with a deficiency in
17α-hydroxylase should hypothetically be divided into the following
three groups: i) Loss of 17α-hydroxylase activity; ii) loss of
17,20-lyase; iii) the combined loss of 17α-hydroxylase/17,20-lyase.
However, it is often difficult to distinguish these forms from
clinical and biochemical results, and a combined deficiency in both
enzymes account for the majority of patients with CAH. In addition,
17,20-lyase deficiency is described in literature less frequently
than 17α-hydroxylase deficiency (11,15).
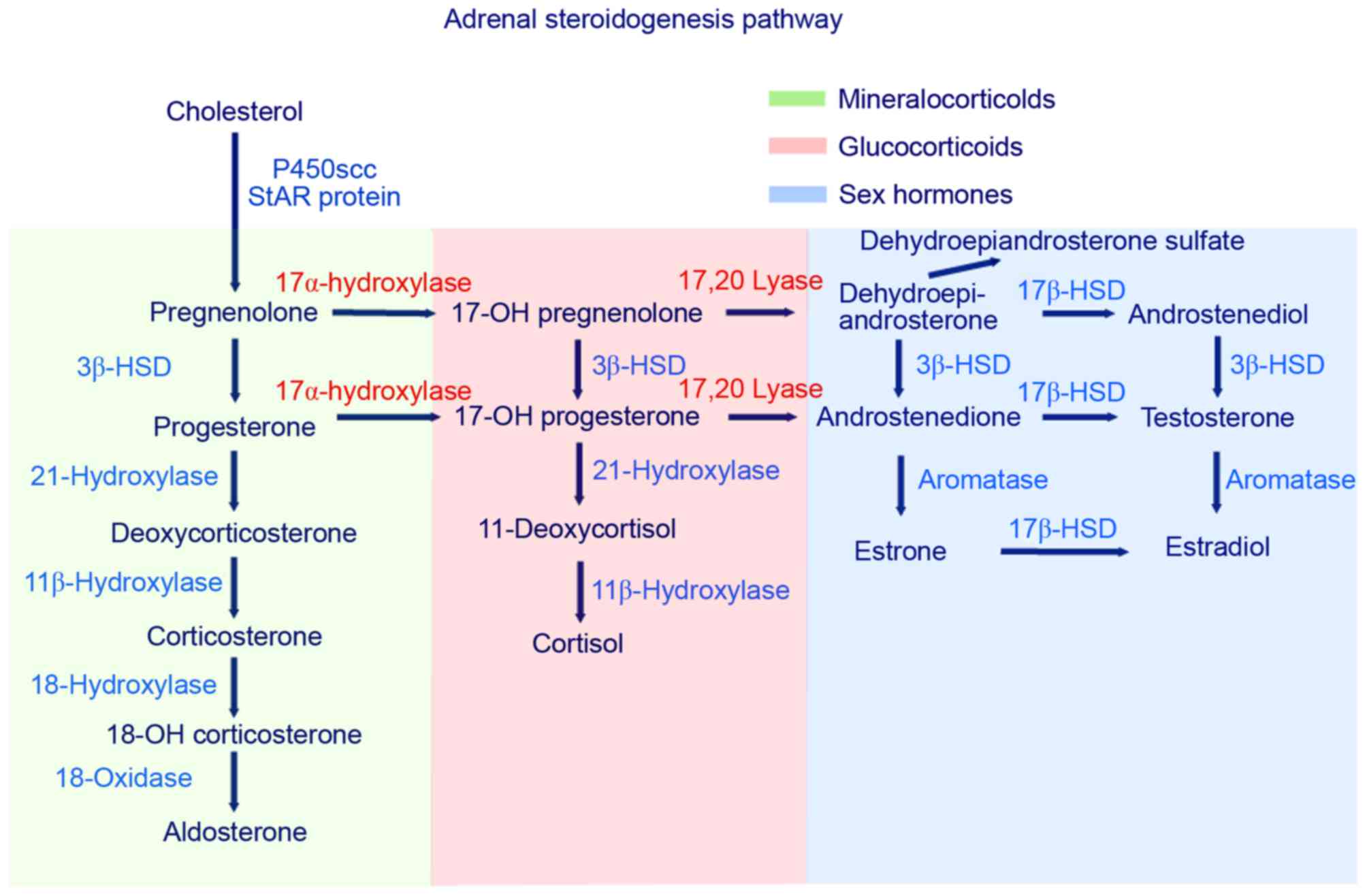
In the steroid biosynthesis pathway,
17α-hydroxylase/17, 20-lyase serves an essential role in cortisol
and sex steroid production (Fig.
2). A deficiency in 17α-hydroxylase/17,20 lyase, caused by a
mutation in the CYP17A1 gene, results in decreased synthesis
of glucocorticoids and androgens (DHEA and androstenedione). In the
biosynthetic pathway, cholesterol is first converted to
pregnenolone. Pregnenolone is then converted to
17α-hydroxypregnenolone by 17α-hydroxylase, leading to its
conversion, via the 3β-hydroxysteroid dehydrogenase pathway, to
progesterone. In patients with CAH that harbor 17,20-lyase
mutations, progesterone cannot be 17α-hydroxylated to
17α-hydroxyprogesterone, as further conversion of these
17α-hydroxylated derivatives to DHEA requires 17,20-lyase. In these
patients, a decrease in 17α-hydroxypregnenolone,
17α-hydroxylprogesterone and DHEA occurs, which leads to a
subsequent reduction in the production of steroids, including
androstenedione, testosterone, 11-deoxycortisol and cortisol.
Accumulation of the substrates, pregnenolone and progesterone,
enhances the 21- and 11β-hydroxylation steps in the
mineralocorticoid pathway, resulting in an increase in
11-deoxycorticosterone, corticosterone and 18-hydroxycorticosterone
levels (1,2,16).
In healthy individuals, decreased cortisol synthesis and the
subsequent feedback inhibition on pituitary ACTH leads to an
increase in ACTH release, in order to return cortisol production to
normal levels. However, in CAH patients there is an overstimulation
of the steroid synthetic pathway, which leads to the overproduction
of mineralocorticoid precursors and hyperplasia of the adrenal
cortex. High levels of mineralocorticoid precursors
(11-deoxycorticosterone, corticosterone and
18-hydroxycorticosterone) induce sodium and fluid retention, and
loss of potassium and hydrogen, which consequently induces
hypertension and hypokalemic alkalosis, due to its potent
mineralocorticoid effect (17).
This feedback suppresses the renin-angiotensin-aldosterone system,
causing hyporeninemic hypoaldosteronism or hyporeninemic
normoaldosteronism (17).
17α-hydroxylase/17,20 lyase and 3β-hydroxysteroid dehydrogenase are
present in the adrenal glands and gonads and contribute to sexual
maturity throughout fetal life and puberty (18). Therefore, there is impairment of
adrenal and gonadal steroid hormone production (androgens and
estrogens) in patients with 17α-hydroxylase/17,20 lyase deficiency.
Affected male individuals have ambiguous or female external
genitalia, however, they present with no internal male genitalia
(prostate, seminal vesicles and vas deferens), which leads to an
absence of testosterone (19).
Female patients present with normal genitalia, but undergo immature
sexual development and develop primary or secondary amenorrhea due
to estrogen deficiency (19).
However, the observed decline in cortisol levels does not manifest
as classical Addison's disease, as raised corticosterone production
demonstrates a mild glucocorticoid effect (10).
Based on the clinical, biochemical and molecular
features, patients may be diagnosed with CAH due to a
17α-hydroxylase/17,20 lyase deficiency. In the current case,
biochemical detection demonstrated that there were decreased
concentrations of estradiol, testosterone, DHEA, androstenedione
and cortisol, as well as increased concentrations of progesterone
and ACTH. The low levels of estradiol and testosterone led to
infantile female genitalia, a lack of secondary sexual
characteristics and primary amenorrhea, as well as an above-average
height. The high levels of FSH and LH, decreased cortisol and
increased ACTH, indicated that primary gonadal failure and primary
adrenal hypocortisolism were evident. Mild hypokalemic alkalosis
may have occurred as a result of mineralocorticoid pathway
activation, which led to renin suppression. However, the patient's
aldosterone levels were within the normal range (10). Notably, the patient exhibited
normotension, which accounts for 10–15% of patients that harbor
mutations in the CYP17A gene in previous reports (10,11).
To the best of our knowledge, ~100 different
CAH-associated mutations that lead to 17α-hydroxylase/17,20-lyase
deficiency have been identified (16). These mutations include missense and
nonsense mutations, insertions, deletions and splice-site variants.
The most common mutation sites differ among ethnic groups, which
can be explained by the founder effect in autosomal recessive
genetic disease (20). The patient
examined in the present study was confirmed to harbor a compound
heterozygous mutation (Y329K418X and D487-S488-F489 deletion) in
the CYP17A1 gene by mutation analysis, which led to the
complete loss of 17α-hydroxylase/17,20-lyase activity. These
mutations have been reported previously in Chinese
17α-hydroxylase/17,20-lyase-deficient patients (21). The c.985–987 mutation (TAC>AA)
has been detected in subjects from Korea and China (initially in a
32-year-old Korean female in 2003) (22), and the c.1459–1467 mutation
(GACTCTTTC deletion) has been detected in subjects from Thailand
and China (initially in a 14-year-old female from Thailand in 1993)
(23). It has been reported that
these mutations in exons 6 and 8 are prevalent in Chinese patients
with 17α-hydroxylase/17,20-lyase deficiency, and may be major
causes of 17α-hydroxylase deficiency in the Chinese population
(21).
Treatments for 17α-hydroxylase/17,20-lyase
deficiency include appropriate glucocorticoid replacement and sex
steroid hormone supplementation. The aim of glucocorticoid
treatment is to reduce ACTH and 11-deoxycorticosterone to normal
levels, using a physiological dose of 0.25–1.0 mg/day dexamethasone
and 2–5 mg/day prednisone. The administration of glucocorticoids
normalizes blood pressure, serum renin and electrolyte levels with
natriuresis. In addition, sex hormone replacement therapy is
recommended to be administered in adolescence for secondary sexual
development, maintenance of female sexual characteristics and
stimulation of epiphyseal closure. Estrogen and progestin cyclic
therapy is required in 46, XX patients to induce cyclic withdrawal
bleeding and prevent endometrial hyperplasia. In addition, estrogen
alone may often be prescribed when patients are admitted at the
time of puberty, at the time of diagnosis in patients that are
postpubescent, or when a male (46, XY) wishes to be considered
female. If a patient decides to be considered male, androgen
replacement therapy is prescribed and extensive genital
reconstructive surgery, such as a gonadectomy to avoid malignant
degeneration in the intra-abdominal testes, may be performed. This
treatment should be discussed with the patient and parents, with an
emphasis on its complexity, implications and consequences (5,24).
In the present case, the patient was administered with prednisone
(5 mg/day) and estradiol valerate (1 mg/day). Despite this sex
steroid replacement, patients with 17α-hydroxylase/17,20-lyase
deficiency develop hypergonadotropic hypogonadism and infertility
due to the decreased enzymatic activity of CYP17A1.
Irreversible defects in steroidogenesis may lead to impaired
spermatogenesis and folliculogenesis, and therefore women are
unable to conceive spontaneously or via endocrinological
intervention (25). However, a
single report of pregnancy in a patient with partial
17α-hydroxylase/17,20-lyase deficiency, resulted in the live birth
of triplets following in vitro fertilization cycles and
transfer of cryopreserved embryos (26). However, to the best of our
knowledge, further attempts at fertility therapy in individuals
with 17α-hydroxylase/17,20-lyase deficiency have not yet resulted
in live births (25).
In conclusion, 17α-hydroxylase/17,20-lyase
deficiency is a rare, unusual and challenging to diagnose endocrine
disorder. This case manifested non-typical clinical symptoms of
17α-hydroxylase/17,20-lyase deficiency (e.g. no
hypertension). To the best of our knowledge the present study was
the first to demonstrate that the two compound heterozygous
mutations in the CYP17A1 gene may be used for the diagnosis
in the 17α-hydroxylase/17,20-lyase deficient patient without
typical clinical symptoms. In the majority of patients, the
presentation of clinical symptoms may be delayed until puberty, and
this subtype of CAH may not be diagnosed at birth. Therefore, some
degree of clinical suspicion is necessary when evaluating children
and adolescents with hypertension and hypokalemia, particularly
when combined with the presence of sexual infantilism, and genetic
analysis is recommended. In the current study, a biochemically and
genetically proven case of 17α-hydroxylase/17,20 lyase deficiency
with 46, XX karyotype was presented. The patient presented with
mild hypokalemia, primary amenorrhea and sexual infantilism, but no
hypertension. Diagnosis of 17α-hydroxylase/17,20 lyase deficiency
was confirmed by the characteristic profile of adrenal steroid
levels, with further confirmation by CYP17A mutation
analysis. Additional analysis of clinical, laboratory and molecular
features may increase our understanding of the CAH endocrine
disease.
Acknowledgments
The authors would like to thank Dr Chen Chen
(Institute of Hypertension and Department of Internal Medicine,
Tongji Hospital, Tongji Medical College, Huazhong University of
Science and Technology, Wuahn, China) for her contribution to
perform genetic analysis of the patient and her family members.
References
|
1
|
Honour JW: Diagnosis of diseases of
steroid hormone production, metabolism and action. J Clin Res
Pediatr Endocrinol. 1:209–226. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Miller WL and Auchus RJ: The molecular
biology, biochemistry, and physiology of human steroidogenesis and
its disorders. Endocr Rev. 32:81–151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krone N and Arlt W: Genetics of congenital
adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab.
23:181–192. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biason-Lauber A, Boscaro M, Mantero F and
Balercia G: Defects of steroidogenesis. J Endocrinol Invest.
33:756–766. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Auchus RJ: The genetics, pathophysiology,
and management of human deficiencies of P450c17. Endocrinol Metab
Clin North Am. 30101–119. (vii)2001.PubMed/NCBI
|
|
6
|
Miller WL: Androgen synthesis in
adrenarche. Rev Endocr Metab Disord. 10:3–17. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miller WL: Steroid 17alpha-hydroxylase
deficiency-not rare everywhere. J Clin Endocrinol Metab. 89:40–42.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oh YK, Ryoo U, Kim D, Cho SY, Jin DK, Yoon
BK, Lee DY and Choi D: 17alpha-hydroxlyase/17, 20-lyase deficiency
in three siblings with primary amenorrhea and absence of secondary
sexual development. J Pediatr Adolesc Gynecol. 25:e103–e105. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller WL: Congenital adrenal
hyperplasias. Endocrinol Metab Clin North Am. 20:721–749.
1991.PubMed/NCBI
|
|
10
|
Kater CE and Biglieri EG: Disorders of
steroid 17 alpha-hydroxylase deficiency. Endocrinol Metab Clin
North Am. 23:341–357. 1994.PubMed/NCBI
|
|
11
|
Yanase T, Simpson ER and Waterman MR: 17
alpha-hydroxylase/17,20-lyase deficiency: From clinical
investigation to molecular definition. Endocr Rev. 12:91–108. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Biglieri EG, Herron MA and Brust N:
17-hydroxylation deficiency in man. J Clin Invest. 45:1946–1954.
1966. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Matteson KJ, Picado-Leonard J, Chung BC,
Mohandas TK and Miller WL: Assignment of the gene for adrenal
P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase) to human
chromosome 10. J Clin Endocrinol Metab. 63:789–791. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Picado-Leonard J and Miller WL: Cloning
and sequence of the human gene for P450c17 (steroid 17
alpha-hydroxylase/17,20 lyase): Similarity with the gene for
P450c21. DNA. 6:439–448. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miller WL: The syndrome of 17,20 lyase
deficiency. J Clin Endocrinol Metab. 97:59–67. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Turcu AF and Auchus RJ: The next 150 years
of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol.
153:63–71. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Goldsmith O, Solomon DH and Horton R:
Hypogonadism and mineralocorticoid excess. The 17-hydroxylase
deficiency syndrome. N Engl J Med. 277:673–677. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Larson A, Nokoff NJ and Travers S:
Disorders of sex development: Clinically relevant genes involved in
gonadal differentiation. Discovery medicine. 14:301–309.
2012.PubMed/NCBI
|
|
19
|
Philip J, Anjali, Thomas N, Rajaratnam S
and Seshadri MS: 17-Alpha hydroxylase deficiency: An unusual cause
of secondary amenorrhoea. Aust N Z J Obstet Gynaecol. 44:477–478.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim YM, Kang M, Choi JH, Lee BH, Kim GH,
Ohn JH, Kim SY, Park MS and Yoo HW: A review of the literature on
common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase
deficiency, a case series of such mutations among Koreans and
functional characteristics of a novel mutation. Metabolism.
63:42–49. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang M, Sun S, Liu Y, Zhang H, Jiao Y,
Wang W and Li X: New, recurrent and prevalent mutations: Clinical
and molecular characterization of 26 Chinese patients with
17alpha-hydroxylase/17,20-lyase deficiency. J Steroid Biochem Mol
Biol. 150:11–16. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hahm JR, Kim DR, Jeong DK, Chung JH, Lee
MS, Min YK, Kim KW and Lee MK: A novel compound heterozygous
mutation in the CYP17 (P450 17alpha-hydroxylase) gene leading to
17alpha-hydroxylase/17,20-lyase deficiency. Metabolism. 52:488–492.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fardella CE, Zhang LH, Mahachoklertwattana
P, Lin D and Miller WL: Deletion of amino acids
Asp487-Ser488-Phe489 in human cytochrome P450c17 causes severe 17
alpha-hydroxylase deficiency. J Clin Endocrinol Metab. 77:489–493.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim SM and Rhee JH: A case of 17
alpha-hydroxylase deficiency. Clin Exp Reprod Med. 42:72–76. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marsh CA and Auchus RJ: Fertility in
patients with genetic deficiencies of cytochrome P450c17 (CYP17A1):
Combined 17-hydroxylase/17,20-lyase deficiency and isolated
17,20-lyase deficiency. Fertil Steril. 101:317–322. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Levran D, Ben-Shlomo I, Pariente C, Dor J,
Mashiach S and Weissman A: Familial partial 17,20-desmolase and
17alpha-hydroxylase deficiency presenting as infertility. J Assist
Reprod Genet. 20:21–28. 2003. View Article : Google Scholar : PubMed/NCBI
|