Introduction
Neuronal cells in the developing brain and central
nervous system are markedly sensitive to anesthetic agents.
Anesthetics are clinical drugs used during surgical and clinical
procedures to induce sedation, and are commonly regarded as safe.
Although the majority of anesthetics have been confirmed to be
safe, some anesthetics exert neurotoxic effects, even at a normal
clinical dose. Contrasting reports exist on the effects of
anesthetics on neuronal physiology and growth. Several anesthetics
and anticonvulsant agents have been reported to exert neuronal
cytotoxicity, neuronal dysfunction, and apoptosis-inducing activity
in vitro and in vivo (1,2). In
previous studies, primary neuronal cells and neonatal mice treated
with ketamine and propofol exhibited blunted dendritic growth,
reduced dendritic spines and arborization (2–4). In
addition, administration of isoflurane to neuronal precursor cells
derived from neonatal rats resulted in a reduced proliferative
capacity (5). Spinal cord neuronal
apoptosis has also been induced by intrathecal administration of
ketamine, but not morphine (6).
Conversely, in other studies, neonatal mice exposed to anesthetics,
including isoflurane, propofol and midazolam, exhibited reduced
neuronal cell death, and dendritic alterations were histologically
improved alongside increased dendritic spine density (7,8).
Furthermore, spinal administration of the anesthetic bupivacaine
has been shown to exert no effect on neuronal apoptosis and
locomotor activity in rats (9).
The immature developing brain passes through various
neurodegenerative processes, including apoptosis, as part of normal
development; however, previous reports have suggested that
anesthetic agents, anticonvulsant drugs and ethanol may accelerate
normal neuronal apoptosis (1–4,6). It
has previously been reported that anesthetics, such as isoflurane
and midazolam, provide protection against neuronal degeneration and
apoptosis, improve histological parameters, and enhance behavioral
and locomotor performance in neonatal rats (2). The dose and duration of anesthetic
exposure has an important role in neuronal histology and cell
growth. Continuous administration of ketamine to rat pups for 9 h
resulted in poor feeding behavior and increased neurodegeneration,
whereas single doses of ketamine exhibited no such effect (10). In addition, propofol infusion may
exert protective effects via effectively reducing hepatic
ischemia/reperfusion injury in rats by decreasing cellular
apoptosis (11). Propofol, with
its antioxidant and anti-inflammatory activity, is considered a
potential hepatoprotective anesthetic in liver surgery. Anesthetics
associated with oxidative stress predominantly induce
Ca2+ release from intracellular stores, including the
rough endoplasmic reticulum (12).
Early indicators of the effects of anesthetics-mediated apoptosis
include reactive oxygen species (ROS) accumulation, mitochondrial
uncoupling and mitochondrial membrane depolarization. These
alterations cause ROS generation, and damage to the mitochondria
and endoplasmic reticulum, thus inducing cell death when
administered in excess.
Midazolam, which is a γ-aminobutyric acid A
(GABAA) receptor agonist of the benzodiazepine class, is
a commonly used anesthetic for the induction of sedation. Midazolam
administration has been shown to preserve dendritic structures, and
does not affect neuronal development during anesthesia (4). Conversely, midazolam activates
apoptosis of cancer cells of various origins, including
hematologic, ectodermal and mesenchymal cells (2,13,14).
Midazolam predominantly acts as an agonist for GABAA
receptor and peripheral-type benzodiazepine receptors (PBRs)
(15,16). PBRs transduce cellular functions,
including cell growth and death, proliferation, and oxidative
processes. The present study investigated the effects of midazolam
on oxidative stress in neuronal cells and elucidated the mechanism
underlying these effects. Midazolam was shown to exert protective
effects against oxidative insults in neuronal cells in vitro
and in vivo via the suppression of ROS and prevention of
neuronal cell death. Therefore, the anesthetic midazolam, with its
antioxidant and anti-apoptotic properties, may be considered a
promising agent in clinical and surgical interventions.
Materials and methods
Preparation of primary cortical
neuronal cell cultures
All animal experimental procedures were performed
after obtaining ethical clearance and in accordance with the
guidelines of the Animal Ethical Committee of the Dongying People's
Hospital (Dongying, China). Primary cortical neuronal cultures were
prepared from mice (obtained from the Beijing Institute of
Laboratory Animals, Chinese Academy of Medical Sciences, Beijing,
China) according to standard protocol. Briefly, mouse neocortices
were obtained from fetal mouse brains between embryonic day 14 and
15. Primary mixed cultures of neuronal and glial cells were
prepared from cerebral cortices newborn mice pups obtained by
mechanical dissociation without trypsin digestion. Neuronal
cultures were grown on cover slips coated with polylysine in
Dulbecco's modified Eagle's medium (Gibco; Thermo Fisher
Scientific, Inc., Inc., Waltham, MA, USA) supplemented with 10%
(v/v) fetal bovine serum (FBS; Thermo Fisher Scientific, Inc.) at
37°C in a CO2 incubator. Neocortices were grown in Eagle's minimum
essential medium (MEM) (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 5% FBS, 5% horse serum (Thermo Fisher Scientific,
Inc.), 21 mM glucose and 2 mM glutamine in a humidified incubator
containing 5% CO2 at 37°C. After 7 days, 10 µM cytosine
arabinofuranoside (Sigma-Aldrich; Merck Millipore, Darmstadt,
Germany) was added to the cell cultures to prevent glial cell
overgrowth. The cells were maintained for 2 weeks for experimental
procedures.
Generation of oxidative stress in
cortical neuronal cells
Cells were maintained as aforementioned, and
oxidative stress was induced as follows: i) Cells were treated with
10 mM buthionine sulfoximine (BSO; Sigma-Aldrich; Merck Millipore)
for 6 h, after which various concentrations of midazolam (25, 50
and 100 µM; Tocris Bioscience, Bristol, UK) were added to the
medium; or ii) cells were treated with 1 mM hydrogen peroxide
H2O2 (Merck Millipore) for 6 h, after which
various concentrations of midazolam were added to the medium. Mixed
cortical cell cultures were then harvested and underwent further
experimentation. Sterile normal buffered saline was used as a
vehicle control throughout in vitro experiments.
Cell viability assay
Cell viability was assessed using the MTT Cell
Proliferation Assay kit (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Cells were treated
with or without midazolam, in combination with either BSO or
H2O2 in 96-well plates. In an experimental
set, cells were pre-treated with 100 µM of trolox (Sigma-Aldrich;
Merck Millipore) for 2 h. Cells were then treated with or without
midazolam, in combination with either BSO or
H2O2 in 96-well plates. After 48 h of
treatment, 20 µl MTT (2 mg/ml) was added to each well and the
plates were incubated for 2 h at 37°C. The MTT-containing medium
was then completely aspirated and 100 µl dimethyl sulfoxide was
added to each well to dissolve the formazan crystals. Subsequently,
absorbance was measured at 570 nm using a microplate reader. The
cell viability ratio was calculated and the value was presented as
a percentage of control.
Determination of intracellular ROS
levels
The production of ROS was measured using the
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA)
assay kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. H2DCFDA is a
chemically-reduced form of fluorescein, which is used as an
indicator of cellular ROS generation. The acetate groups are
cleaved by intracellular esterases and oxidation, and the
non-fluorescent H2DCFDA is converted to the highly
fluorescent 2′,7′-dichlorofluorescein (DCF). Briefly, the cortical
neuronal cells were treated with BSO or H2O2
with or without midazolam for 24 h. Cells were then incubated with
10 mM H2DCFDA for 30 min at 37°C in the dark. The cells
were rinsed twice with fresh MEM supplemented with 1% FBS, and were
washed three times with cold PBS. Subsequently, cells were fixed
with 4% paraformaldehyde in PBS for 5 min at room temperature,
washed twice with PBS and sealed with mounting media containing
4′,6-diamidino-2-phenylindole (DAPI; Abcam, Cambridge, MA, USA).
Images of the cells were captured using laser scanning confocal
microscopy (Nikon Corporation, Tokyo, Japan).
Protein isolation and western
blotting
Cell cultures were treated as aforementioned in
6-well plates prior to protein isolation. After treatment, cells
were harvested, washed twice with ice-cold PBS and pelleted by
centrifugation at 2,000 × g for 2 min at 4°C. The cell
pellets were lysed using cell lysis buffer containing 20 mM Tris
(pH 7.5), 1 mM EDTA, 150 mM NaCl, 2.5 mM sodium pyrophosphate, 1%
Triton X-100, 1% sodium vanadate, 1 mM phenylmethylsulfonyl
fluoride and protein inhibitor cocktail (Invitrogen; Thermo Fisher
Scientific, Inc.). The cells were incubated at 4°C for 30 min with
intermittent vortex mixing. Subsequently, lysates were centrifuged
at 10,000 × g for 10 min at 4°C and the supernatants were
collected. Protein concentrations were estimated from the
supernatants using the bicinchoninic acid protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.). Equal amounts of protein
(20 µg) in each sample were separated by 12% SDS-PAGE and proteins
were transferred onto polyvinylidene fluoride transfer membranes
(EMD Millipore, Billerica, MA, USA). Membranes were blocked in
fat-free skimmed milk at room temperature for 2 h and were then
washed twice with TBS-1% Tween (TBST). Subsequently, membranes were
incubated with protein-specific primary antibodies at 4°C
overnight, after which they were washed three times with TBST at
room temperature and were incubated with anti-mouse (cat. no. 7076)
and anti-rabbit (cat. no. 7074) horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000; Cell Signaling
Technology, Inc., Danvers, MA, USA) for 90 min at room temperature.
The blots were visualized using enhanced chemiluminescence solution
(Invitrogen; Thermo Fisher Scientific, Inc.). All protein bands
were semi-quantified using a densitometer and were analyzed using
Bio-Rad Image Analysis Software version 4.1 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). Primary antibodies used were as follows:
Anti-caspase-3 (1:1,000; cat. no. 6992), anti-caspase-9 (1:1,000;
cat. no. 9504), anti-poly (ADP-ribose) polymerase (PARP; 1:1,000;
cat. no. 9542), anti-BH3 interacting-domain death agonist (Bid;
1:1,000; cat. no. 2003), anti-B-cell lymphoma 2 (Bcl2; 1:1,000;
cat. no. 3498), anti-phosphorylated (p)extracellular
signal-regulated kinases (ERK; 1:1,000; cat. no. 9101), anti-AKT
(1:1,000; cat. no. 9272), anti-pAKT (1:2,000; cat. no. 4060),
anti-c-Jun N-terminal kinases (JNK; 1:1,000; cat. no. 3708) and
anti-pJNK (1:1,000; cat. no. 4668; Cell Signaling Technology,
Inc.); anti-pERK (1:2,000; cat. no. sc-7383), anti-GAPDH (1:2,000;
cat. no. sc-32233) and anti-protein kinase C (PKC)-ε (1:1,000; cat.
no. sc-1681; Santa Cruz Biotechnology, Inc., Dallas, TX, USA); and
anti-nuclear factor (NF)-κB (p65; 1:1,000; cat. no. NB100-2176;
Novus Biologicals LLC, Littleton, CO, USA).
Middle cerebral artery occlusion
(MCAO) in mice and midazolam administration
Female BALB/c mice (age, 5 weeks; weight, 20–24 g)
were housed under standard laboratory conditions at 22–24°C with
humidity at 50–60% and a 12-h light/dark cycle. Mice were allowed
access to food and water ad libitum. The mice were randomly
divided into four groups (n=20/group): Control group; MCAO group;
MCAO + midazolam treatment group-1 (2 mg/kg); and MCAO + midazolam
treatment group-2 (5 mg/kg). Midazolam was administered
subcutaneously (s.c.) prior to MCAO. Transient MCAO in mice was
induced according to the standard intraluminal suture method
(17). Briefly, mice were
anesthetized using 50 mg/kg pentobarbital sodium. A nylon
monofilament with a silicone-beaded tip was inserted into the right
internal carotid artery of the mice through the external carotid
artery to occlude the origin of the middle cerebral artery.
Reperfusion (24 h) was performed after 90 min of occlusion.
Polyethylene catheters inserted into the right femoral artery were
used for the measurement of blood pressure, and for the detection
of arterial blood gases and glucose sampling. The body temperature
of the mice during surgical procedures was maintained at 37°C
(±0.5°C) with the use of a heating pad.
Neurological deficits analysis
Neurological examination was performed after 24 h of
ischemia/reperfusion in mice. Neurological deficits in mice
(n=8/group) were assessed using a standard 5-point scale as
described previously (18). The
scale was based on the following deficit criteria: 0, no deficit;
1, failure to fully extend left forepaw; 2, circling to the left;
3, falling to the left; 4, unable to walk spontaneously or
stroke-associated mortality. The examination was performed by an
investigator that was blinded to the animal groupings.
Measurement of infarct size and
volume
After neurological testing, mice were anesthetized
with pentobarbital sodium (50 mg/kg) and were decapitated. Brains
were collected from the rats (n=5/group) and were sliced into five
coronal sections (1 mm). The brain sections were stained with 2%
2,3,5-triphenyltetrazolium chloride at 37°C for 30 min and were
fixed in 4% paraformaldehyde. Imaging of the stained cerebral
sections was performed and analyzed using the Image Pro-Plus
analysis system version 6.3 (Media Cybernetics, Inc., Rockville,
MD, USA). Infarct size was determined using the following equation:
Corrected infarct volume = contralateral hemisphere volume -
(ipsilateral hemisphere volume - measured infarct volume). Infarct
volume was presented as the percentage in the brain samples
compared with the control. Infarct volume measurements were
performed by an investigator that was blinded to the animal
groupings.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) apoptosis assay
The TUNEL assay was performed using the In
Situ Cell Death Detection kit (Roche Diagnostics, Minneapolis,
MN, USA) according to the manufacturer's protocol. After
neurological testing, mice (n=3/group) were anesthetized with
pentobarbital sodium (50 mg/kg) and were decapitated.
Paraffin-embedded sections were deparaffinized in xylene and
rehydrated in a graded ethanol series to water. Excess water was
removed from slides and sections were permeabilized with 20 µg/ml
proteinase K (Gibco; Thermo Fisher Scientific, Inc.) prepared in 10
mM Tris buffer (pH 7.5) containing 5 mM EDTA. Sections were
incubated at room temperature for 30 min and subsequently treated
with 3% hydrogen peroxidase for 30 min and washed in PBS to quench
endogenous peroxidases. Following equilibration, the sections were
end labeled with digoxigenin-11-deoxyuridine triphosphate by
incubation with the terminal deoxynucleotidyl transferase enzyme in
buffer containing 50 mM potassium acetate, 20 mM Tris-acetate, 10
mM magnesium acetate (pH 7.9) supplemented with 0.25 mM CoCl2 for 1
h at 37°C in a humidified chamber. Following treatment with
stop-wash buffer, the sections were incubated with DAPI for 30 min
and TUNEL-positive cells and double stained cells of different
groups were observed randomly in the penumbra under a light
microscope.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Tissue was homogenized at 4°C and total RNA was
extracted from brain sections (n=3/group) using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. Total RNA was quantified
spectrophotometrically and 1 µg RNA was reverse transcribed using
SuperScript III kit (Invitrogen; Thermo Fisher Scientific, Inc.).
Briefly, RNA was added to 0.5 mM dNTPs, 200 ng random hexamers,
reaction buffer containing 5 mM DTT, 40 units RNAse A and 200 units
reverse transcriptase, and was processed in a thermal cycler at
65°C for 30 min. The obtained cDNA was used for differential PCR
amplification of targeted genes in an ABI PRISM 7500 system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Target
amplification was performed using TaqMan minor groove binder probes
labeled with fluorescein dye (Applied Biosystems; Thermo Fisher
Scientific, Inc.) in a 20 µl reaction mixture containing 2 µl cDNA,
ROX passive reference dyes and TaqMan Universal PCR mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling
conditions were as follows: 95°C for 5 min; followed by 35 cycles
of 95°C for 10 sec, 60°C for 20 sec and 72°C for 15 sec. The
specific primer sets used were as follows: Parp1 (Entrez Gene ID,
25591; Rn00565018_m1) and caspase-3 (Casp3) (Entrez Gene ID, 25402;
Rn00563902_m1) (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The human 18S rRNA was used as an internal control (Entrez
Gene ID, 19791; NR_003278), and for normalization of the relative
quantification of target gene expression. The relative
quantification was performed using the 2−∆∆Cq method as
previously described (19).
Neuroapoptotic mice and midazolam
administration
Female BALB/c mice (age, 7 days; weight, 2.2–2.5 g)
were used for analyzing neuroapoptosis due to their sensitivity to
anesthetic agents, such as GABAA or N-methyl-D-aspartate
agonists (14). The mice were
housed at 22–24°C with humidity at 50–60% and a 12-h light/dark
cycle. Mice were allowed access to food and water ad
libitum. The mice were randomly divided into four groups
(n=10/group): Control group; ethanol group (5 mg/kg); ethanol +
midazolam treatment group-1 (2 mg/kg); ethanol + midazolam
treatment group-2 (5 mg/kg). Neuroapoptosis was induced in the
brains of the mouse pups via administration of ethanol, which is a
neurotoxic agent to the developing brain, according to a previously
described method (20). Mice in
the control group received normal saline (s.c.). The total volume
of administered solutions was <50 µl per mouse. The body
temperature of the mice was maintained at 37°C (±0.5°C) with the
use of a heating pad. Mice were initially administered midazolam
for 6 h, followed by a single injection of ethanol, and were then
returned to normal conditions. A total of 6 h after ethanol
administration, the mice were anesthetized with 2.5% isoflurane and
decapitated, and the brains were extracted and fixed in
paraformaldehyde.
Immunohistochemistry (IHC) and TUNEL
staining for apoptosis
Paraffin-embedded tissues were prepared by standard
method. Brain samples were extracted, washed with PBS then fixed in
4% paraformaldehyde in PBS for 6 h. Following fixation, tissues
were rinsed with PBS to completely remove fixative. Tissues were
subsequently dehydrated in graded alcohol and subjected to paraffin
infiltration in molten paraffin wax at 65°C in blocks. The wax
infiltrated tissues, embedded in blocks, were subjected to further
procedure. Brain histology was studied in the paraffin-embedded
sections (n=5/group). Briefly, brain sections were deparaffinized
and antigen retrieval was performed using Dako Target Retrieval
Solution (Dako Denmark A/S, Glostrup, Denmark) according to the
standard protocol. Tissue sections were washed with water and
incubated with 3% H2O2 for 5 min to block endogenous peroxidase.
The sections were then incubated with anti-active caspase-3 (cat.
no. 9661; Cell Signaling Technology, Inc.) diluted to 1:500 in
blocking agent, for 30 min at room temperature. Subsequently,
tissue sections were washed with TBST and incubated with
HRP-labeled anti-rabbit secondary antibody (1:500; cat. no. E0432;
Dako Denmark A/S) for 30 min at room temperature followed by
visualization with 3,3′-diaminobenzidine chromogen (Dako Denmark
A/S). Counterstaining was performed with hematoxylin, and
caspase-3-positive staining was documented under a light
microscope. The scoring of caspase-3-positive cells per
mm2 brain section was performed by an investigator that
was blinded to the animal groupings. The total number of
caspase-3-positive cells was presented as a percentage of positive
cells compared to the control group. TUNEL staining of the brain
sections (n=5/group) was conducted as aforementioned.
Statistical analyses
Experiments were repeated in triplicate. Data are
presented as the mean ± standard deviation and statistical
differences among groups were calculated using Student's t-test or
one-way analysis of variance followed by Tukey's post-hoc test.
Neurological deficit was analyzed by Kruskal-Wallis test with
Bonferroni correction. Statistical analyses were conducted using
SPSS v.16.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significance difference.
Results
Effects of midazolam on oxidative
stress-induced neuronal cell death
In order to investigate the effects of midazolam on
neuronal cell function and physiology, the present study induced
oxidative stress in murine cortical neuronal cells. Two oxidative
stress inducers (BSO and H2O2) were
individually used to analyze the effects of midazolam. BSO is a
sulfoximine compound that reduces glutathione levels by inhibiting
γ-glutamylcysteine synthetase, which is the enzyme essential for
the first step of glutathione synthesis. BSO induces generation of
intracellular ROS and ultimately causes apoptosis (21). H2O2 is
another inducer of intracellular ROS generation in neuronal cells
(22). In the present study,
primary cortical neuronal cell cultures were treated with BSO or
H2O2 and were then treated with or without
midazolam for 48 h prior to assessment of cell viability. The
results of an MTT assay demonstrated that BSO (10 mM) inhibited the
growth of cortical neuronal cells to 61% compared with the control
cells. Treatment of cells with midazolam increased cell growth in a
dose-dependent manner; cells treated with 100 µM midazolam
exhibited 82% cell viability, as compared with the control cells
(Fig. 1A). In cells treated with
H2O2, H2O2 (1 mM)
inhibited the growth of cortical neuronal cells to 48% compared
with the control cells. Treatment of cells with midazolam increased
cell growth in a dose-dependent manner; cells treated with 100 µM
midazolam exhibited 78% cell survival, as compared with the control
cells (Fig. 1B). Midazolam (100
µM) alone was found to be non-cytotoxic to cortical neuronal cells;
cells treated with 100 µM midazolam exhibited 94 and 96% cell
viability compared with the control cells. The protective effects
of midazolam in oxidative stress-induced cortical neuronal cells
were further confirmed by western blotting of apoptosis-associated
proteins (Fig. 1C). BSO treatment
(10 mM) resulted in activation of pro-caspase-3 and cleavage of
PARP. However, 100 µM midazolam treatment resulted in reduced
levels of activated caspase-3 and cleaved-PARP (Fig. 1C). Furthermore, BSO induced
truncation-activation of Bid and reduced Bcl2 expression; these
effects were reversed by treatment with midazolam (Fig. 1C). Similarly,
H2O2 treatment (1 mM) resulted in activation
of pro-caspase-3, cleavage of PARP, truncation-activation of Bid
and reduced expression of Bcl2. Conversely, midazolam treatment
attenuated the effects of H2O2 (Fig. 1C). Activation of pro-caspase-9 in
both oxidative stress conditions indicates the putative activation
of the mitochondrial intrinsic apoptosis pathway, which was
suppressed following exposure to midazolam. These results suggest
that BSO and H2O2 may inhibit the
proliferation of cortical neuronal cells by inducing apoptosis, and
that the treatment of cells with midazolam may recover cell
growth.
Midazolam suppresses generation of
intracellular ROS in neuronal cells
The effects of midazolam on intracellular ROS levels
were determined by H2DCFDA assay. Treatment of cortical
neuronal cells with BSO (10 mM) and H2O2 (1
mM) resulted in increased production of intracellular ROS, as
demonstrated by increased cellular fluorescence intensity (Fig. 2A). Quantification of intracellular
ROS levels was determined by measuring DCF intensity. Cortical
neuronal cells treated with BSO (10 mM) exhibited increased DCF
intensity (154±12%) compared with the control cells (100%).
BSO-induced DCF intensity was subsequently inhibited following
treatment with 100 µM midazolam (122±11%). Similarly,
H2O2 (1 mM) treatment increased DCF intensity
to 178±11% compared with the control group; however, DCF intensity
was subsequently inhibited following treatment with 100 µM
midazolam (132±12%) (Fig. 2B).
These results indicate that midazolam has the ability to suppress
intracellular ROS accumulation, and may possess free radical
scavenging activities, thus preventing neuronal cell death.
Furthermore, the present study detected the viability of oxidative
stress-induced cortical neuronal cells following treatment with
midazolam in combination with trolox. Trolox
(6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) is a
well-known antioxidant that suppresses intracellular ROS
generation. In the BSO-treated groups, cell viability percentages
for each group were as follows: BSO (10 mM), 64%; trolox (100 µM),
74%; and midazolam (100 µM), 80% (Fig.
2C). The combination of trolox and midazolam exerted a
synergistic effect; cell viability was 92% compared with the
control cells (100%). In the H2O2-treated
groups, the cell viability percentages for each group were as
follows: H2O2 (1 mM), 51%; trolox (100 µM),
71%; and midazolam (100 µM), 75% (Fig.
2D). The combination of trolox and midazolam exerted a
synergistic effect; cell viability was 88% compared with the
control cells. Collectively these data indicate that BSO and
H2O2 induce oxidative stress in cortical
neuronal cells resulting in growth inhibition. The reversal of BSO
and H2O2-induced effects by midazolam
indicates its ROS inhibitory properties.
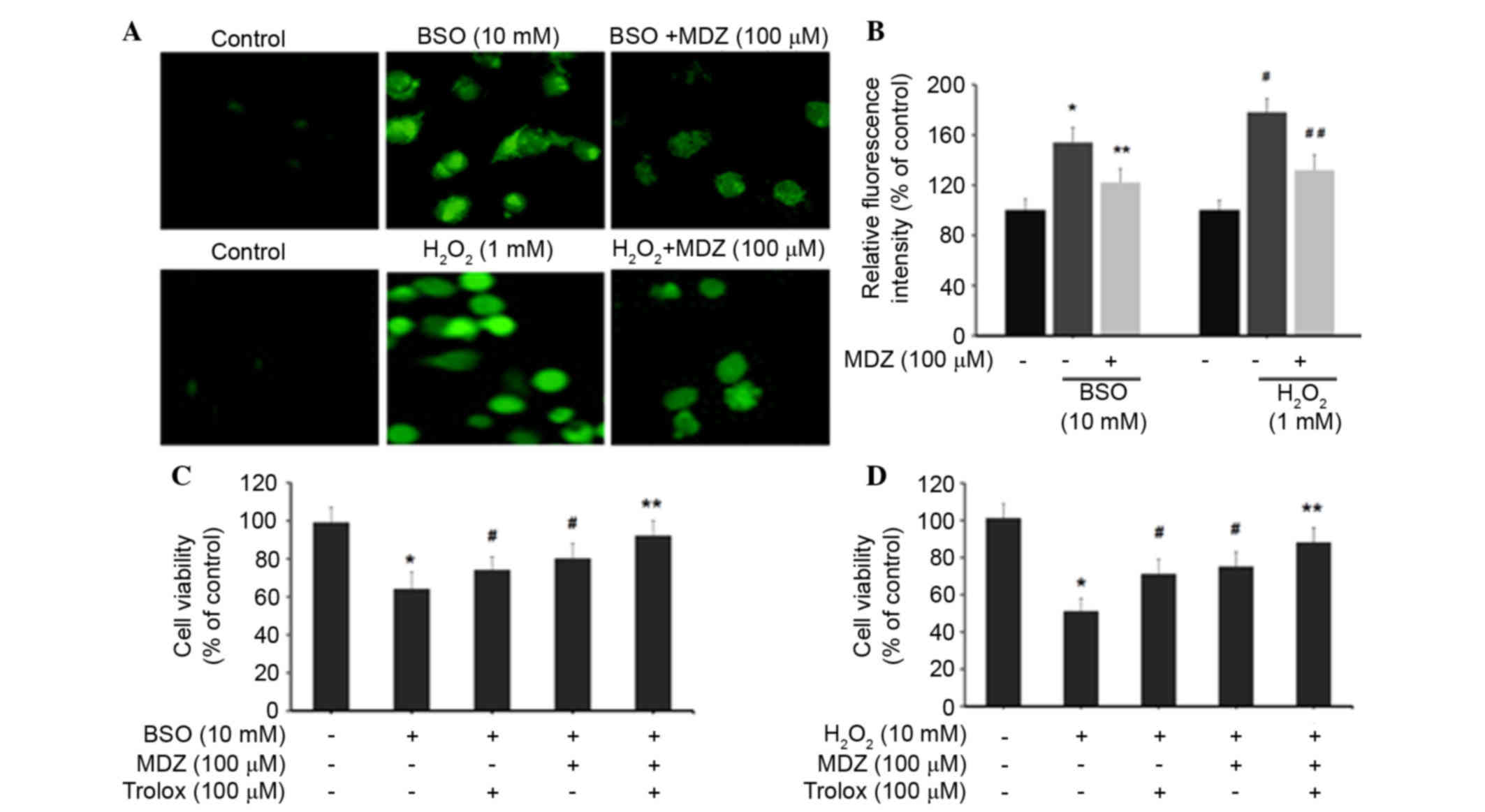 | Figure 2.Effects of midazolam on intracellular
ROS generation in oxidative stress-induced cortical neuronal cells.
(A) ROS generation was measured using the H2DCFDA assay.
Cortical neuronal cells were treated with
BSO/H2O2 and midazolam for 24 h. Cells were
then incubated with 10 mM H2DCFDA and images were
captured using laser scanning confocal microscopy. (B)
2′,7′-dichlorofluorescein intensity was quantified and represented
as relative fluorescence intensity (percentage of control).
*P<0.05 vs. the control, **P<0.05 vs. BSO,
#P<0.05 vs. the control, ##P<0.05 vs.
H2O2. (C and D) Cells were treated with
BSO/H2O2 in combination with midazolam and
trolox, and cell viability was assessed. Data are presented as the
mean ± standard deviation. *P<0.05 vs. the control,
#P<0.05 vs. BSO/H2O2, **P≥0.05
vs. the control. BSO, buthionine sulfoximine;
H2O2, hydrogen peroxide; MDZ, midazolam;
H2DCFDA, 2′,7′-dichlorodihydrofluorescein diacetate. |
Midazolam enhances survival in
cortical neuronal cells under oxidative stress
The present study aimed to elucidate the molecular
mechanisms underlying the protective effects of midazolam on
oxidative stress-induced apoptosis in cortical neuronal cells.
Western blotting demonstrated that treatment with BSO and
H2O2 suppressed the expression levels of cell
survival-associated proteins in cortical neuronal cells (Fig. 3). BSO (10 mM) and
H2O2 (1 mM) suppressed the protein expression
levels of ERK1/2, pERK1/2, AKT and pAKT. In BSO and
H2O2 treatment groups, midazolam (100 µM)
recovered the protein expression levels of ERK, pERK, AKT and pAKT.
Suppression of pERK and pAKT by BSO and H2O2
was comparatively higher compared with the suppression of their
native forms. In addition, BSO and H2O2
suppressed activation of JNK and pJNK proteins, which was reversed
following treatment with midazolam (100 µM). Suppression of JNK and
pJNK activation by BSO and H2O2 is directly
associated with the suppression of major cell survival signals in
cortical neuronal cells. Furthermore, the recovery of JNK and pJNK
expression by midazolam indicates its protective effects against
BSO and H2O2. BSO and
H2O2 also suppressed the expression of NF-κB,
which is transcriptional regulator of the Bcl-2 family that
contains pro- and anti-apoptosis genes. This family of genes also
regulates cell survival signaling. Thus, an increase in NF-κB by
midazolam results in an increase in cell survival, suggesting
reduced apoptosis. Midazolam was shown to elevate the levels of
NF-κB, which were inhibited by BSO and H2O2
(Fig. 3). NF-κB modulation is also
associated with ERK and AKT signaling, as corroborated by the
present results. These results suggest that the protective effects
of midazolam on oxidative stress-induced cortical neuronal cells
are mediated via modulation of JNK and NF-κB signaling. BSO has
been reported to act through inhibition of PKC cell signaling
pathway proteins, particularly PKC-ε (21). Therefore, the present study
analyzed the protein expression levels of PKC-ε in BSO and
midazolam-treated cortical neuronal cells by western blotting. As
hypothesized, BSO (10 mM) inhibited the protein expression levels
of PKC-ε, whereas midazolam (100 µM) treatment moderately recovered
PKC-ε expression. In addition, trolox suppressed the effects of BSO
by elevating PKC-ε levels. The combination of trolox and midazolam
exerted a synergistic effect on the recovery of PKC-ε protein
levels, which were inhibited by BSO (Fig. 3). These results provide information
regarding the mechanism underlying midazolam-induced suppression of
oxidative stress in cortical neuronal cells.
Effects of midazolam on neuronal
deficit and infarct volume in MCAO mice
In order to investigate the effects of midazolam on
stress and ischemia, MCAO mice were used. Proximal occlusion of the
middle cerebral artery is one of the most frequently used
experimental models of stroke. The MCAO model is among the most
common models of ischemic stroke and stress in the developing brain
(17). MCAO mice are characterized
by neuronal deficit, neuronal degeneration and cell death. In the
present study, neuronal deficits in mice were evaluated 24 h
following transient MCAO reperfusion (Table I). The results demonstrate that
transient MCAO reperfusion induced severe neuronal deficits in
mice; with 50% of mice classified as grade 3 and 12.5% of mice
classified as grade 4, as compared with the control group.
Midazolam post-conditioning in MCAO mice improved the neurological
outcomes in a dose-dependent manner. Neuronal deficits in MCAO mice
were improved following treatment with 2 mg/kg midazolam, with a
median value of 2 (P<0.042) and with 5 mg/kg midazolam with a
median value of 4 (P<0.028), as compared with the MCAO group
(median value of 1). These results indicate that midazolam may
exert protective effects against neuronal deficit and degeneration
in mice with transient intraluminal occlusion. In addition, the
present study examined the effects of midazolam on infarct size in
MCAO mice. Brain sections were analyzed to determine the percentage
of infarct area in the whole brain, and the results were presented
as a percentage of the control (Fig.
4). Mice in the MCAO group exhibited extensive infarction
(52±7%) compared with the control group. Conversely, midazolam
post-conditioning in MCAO mice significantly reduced infarct volume
in a dose-dependent manner. Treatment of MCAO mice with 2 or 5
mg/kg midazolam reduced infarct size to 42±7% (P<0.05) and 34±6%
(P<0.03), respectively, as compared with the control group.
These data suggest that midazolam exerts protective effects on
MCAO-induced brain infarction in mice.
 | Table I.Neurological deficit scoring
following ischemia/reperfusion (n=8/group). |
Table I.
Neurological deficit scoring
following ischemia/reperfusion (n=8/group).
|
| Neurological
deficit scores |
|
|---|
|
|
|
|
|---|
| Group | 0 | 1 | 2 | 3 | 4 | Median (range) |
|---|
| Control | 8 | 0 | 0 | 0 | 0 | 0 |
| MCAO | 0 | 1 | 2 | 4 | 1 | 1 (1–4) |
| MCAO + MDZ (2
mg/kg) | 1 | 3 | 2 | 2 | 0 | 2
(0–3)a |
| MCAO + MDZ (5
mg/kg) | 3 | 4 | 1 | 0 | 0 | 4
(0–4)b |
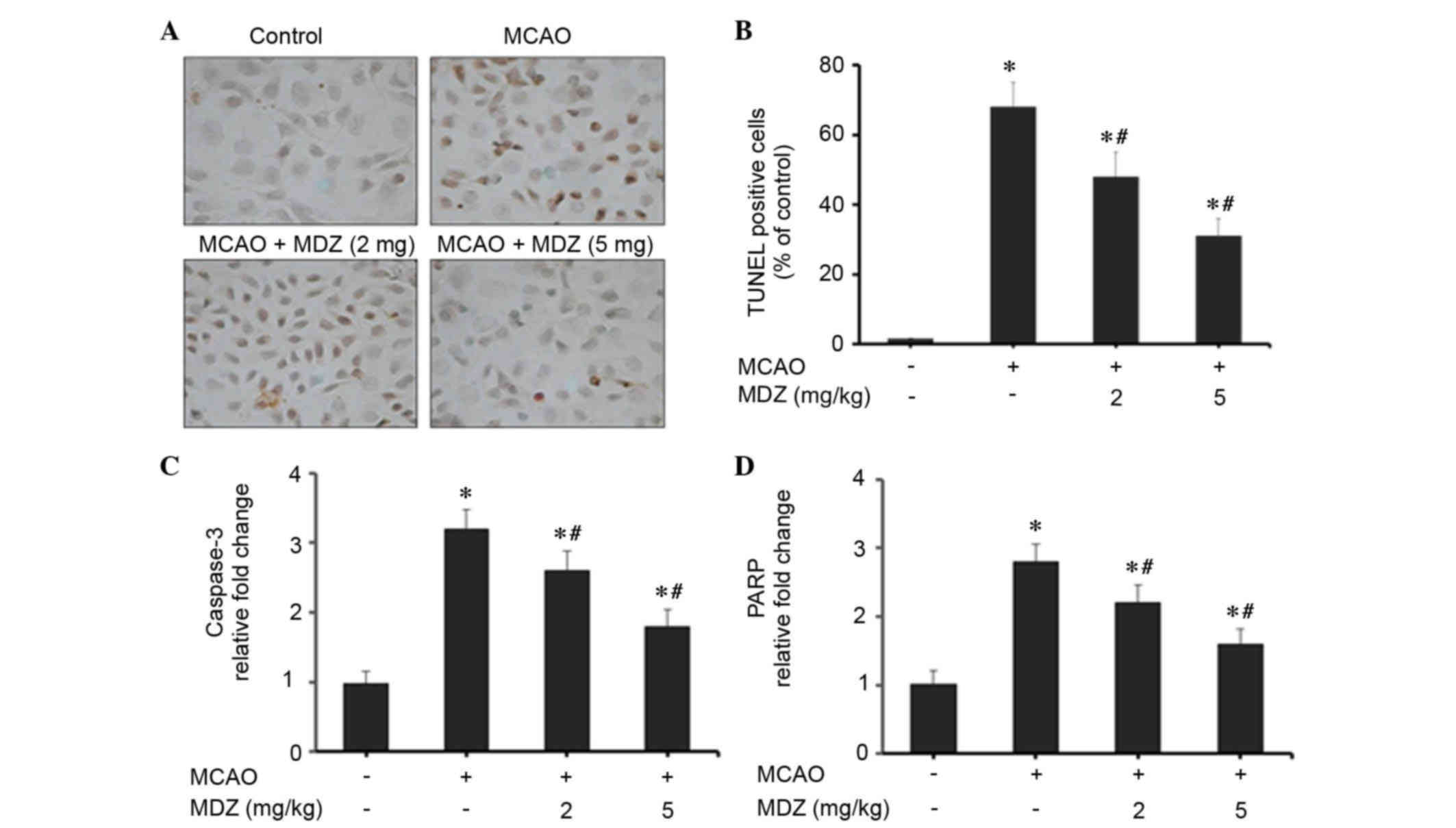
Midazolam suppresses neuronal
apoptosis in MCAO mice
The present study examined the effects of midazolam
on neuronal apoptosis in MCAO mice using TUNEL staining and RT-qPCR
of apoptosis-associated genes (Fig.
5). Transient MCAO and reperfusion increased cellular apoptosis
in the brain cortex compared with in the control group; however,
apoptosis was markedly suppressed following treatment with
midazolam in a dose-dependent manner (Fig. 5A). The number of cells stained for
apoptotic bodies was markedly increased in the MCAO group compared
with in the control group. Midazolam-treated MCAO mice exhibited a
reduction in the number of apoptotic cells compared with the MCAO
group. As presented in Fig. 5B,
quantitative scoring of TUNEL-positive cells was conducted; a high
percentage of TUNEL-positive apoptotic cells was detected in the
MCAO group (68±7%). However, treatment of MCAO mice with 2 or 5
mg/kg midazolam reduced the percentage of TUNEL-positive apoptotic
cells to 48±7% (P<0.04) and 31±5% (P<0.03), respectively.
These findings emphasize the anti-apoptotic properties of midazolam
in MCAO mice. The effects of midazolam on apoptosis in MCAO mice
were further confirmed by RT-qPCR analysis of apoptosis-associated
genes (caspase-3 and PARP). The mitochondrial intrinsic apoptosis
pathway induces cell death via activation of the executioner
caspase-3, followed by cleavage of the nuclear substrate PARP. In
MCAO mice, the expression levels of caspase-3 and PARP were
increased 3.2- and 2.8-fold, respectively, as compared with in the
control group (Fig. 5C and D). The
expression levels of caspase-3 and PARP were dose-dependently
suppressed following treatment of MCAO mice with midazolam.
Treatment with 2 or 5 mg/kg midazolam reduced the expression levels
of caspase-3 to 2.6- and 1.8-fold, respectively, as compared with
the control group (Fig. 5C).
Similarly, treatment with 2 or 5 mg/kg midazolam reduced the
expression levels of PARP to 2.2- and 1.6-fold, respectively,
compared with the control group (Fig.
5D). These findings were statistically significant when
compared with the control group. Collectively, these data suggest
that the protective effects of midazolam are attributed to its
anti-apoptotic activities in MCAO mice.
Effects of midazolam on
ethanol-induced neuronal apoptosis in mice
Ethanol exposure to infant rodents during neuronal
development, particularly rapid synaptogenesis, can induce
extensive neuronal apoptosis and degeneration (20). In the present study, the effects of
midazolam on a mouse model of ethanol-induced neurodegeneration and
apoptosis were assessed by activated caspase-3 IHC and TUNEL
staining. The quantitative estimations of apoptosis were recorded
and presented as a percentage of the control (Fig. 6). Quantification of the IHC results
indicate that ethanol induced severe apoptosis in the brains, as
demonstrated by the increased number of caspase-3-positive cells
compared with the control group. Ethanol-induced a 4.8-fold
increase in caspase-3-positive cells compared with the control
(Fig. 5A). Conversely, treatment
with midazolam resulted in a dose-dependent suppression in
caspase-3-positive cells; treatment with 2 or 5 mg/kg midazolam
suppressed the number of caspase-3-positive cells to 3.4- and
2.4-fold, respectively, as compared with the control group
(Fig. 6A). The effects of
midazolam on ethanol-induced neurodegeneration and apoptosis were
further confirmed by TUNEL staining of the brain cortex. TUNEL
staining of apoptotic cells was recorded as a percentage of the
control (Fig. 6B). Exposure of
mice to ethanol resulted in a marked increase in the percentage of
TUNEL-positive cells (58±9%) compared with the control group.
Treatment of ethanol-exposed mice with midazolam resulted in a
dose-dependent suppression in TUNEL staining. Treatment with 2 or 5
mg/kg midazolam suppressed the ratio of TUNEL-positive cells to 43
and 32%, respectively, as compared with the control group (Fig. 6B). These data indicate that
midazolam protects the developing mouse brain from ethanol-induced
neurodegeneration and apoptosis.
Discussion
Anesthetic and anticonvulsant agents are commonly
used to induce sedation during clinical and surgical procedures.
Although several such agents have been shown to induce neuronal
degeneration, cytotoxicity and dysfunction, some have been reported
to exert protective properties. The present study demonstrated that
the commonly used anesthetic midazolam exerts protective effects on
neuronal cells and the developing brain under physiological and
oxidative stress conditions. Transient occlusion-induced apoptotic
neuronal degeneration and ethanol-induced neuroapoptosis in the
murine brain was reversed following treatment with midazolam.
Furthermore, midazolam recovered oxidative stress-inhibited
cortical neuronal cell growth via suppression of ROS, and
modulation of JNK-ERK and NF-κB signaling pathways.
Ischemia/reperfusion in the developing brain may
induce severe neurological damage, which may trigger a complex
series of biochemical events resulting in structural and functional
deficits in the nervous system. Transient global ischemia initiates
intricate neuronal cell death processes following ischemic insult,
particularly in developing brains. Anesthetics are used for their
sedative properties; however, contradictory neurocytotoxic and
neuroprotective properties have been reported in various in
vitro and in vivo systems (1,2,11,23,24).
Recently, propofol was demonstrated to exert neuroprotective
effects in hypoxia-induced hippocampal neuronal injury; it improved
alterations in neuronal structure and decreased the release of
amino acid neurotransmitters in hypoxic brains (25). Furthermore, propofol exerted
neuroprotective effects in brain ischemia/reperfusion injury via
the induction of heme oxygenase-1 expression (26). Propofol has also been reported to
attenuate activation of caspase-3 in the cortex and hippocampus of
mice, and may improve cognitive function, possibly through
GABAA receptor action (27). In addition, isoflurane and
midazolam have been demonstrated to be protective against neuronal
degeneration and apoptosis (2).
Another protective mechanism in neuronal cells is mediated by the
suppression of oxidative stress-induced ROS. The natural polyphenol
quercetin has been demonstrated to protect neuronal cells from
oxidative stress-induced apoptosis by suppressing ROS accumulation
and activation of the JNK, ERK1/2 and Akt signaling pathways
(28). Resveratrol and quercetin
also exert protective effects against oxidative stress-induced
apoptotic cell death in dopaminergic neurons by suppressing ROS and
modulating apoptotic markers (29). Therefore, clinical and therapeutic
interventions that suppress oxidative stress may effectively
protect against neuronal damage and neuroapoptosis.
The present study investigated the effects of
midazolam, a rapidly acting intravenous hypnotic agent, on neuronal
apoptosis and neurodegeneration. The results suggested that
midazolam may improve neurological outcome in animal models of
cerebral ischemia and neuroapoptosis. The neuroprotective effects
of midazolam were comprehensively characterized in cortical
neuronal cells, and in murine models of MCAO ischemia/reperfusion
injury and ethanol-induced neuroapoptosis. BSO (10 mM) and
H2O2 (1 mM) were used to induce oxidative
stress in primary cortical neuronal cells prepared from the
developing mouse brain. BSO and H2O2
suppressed the proliferation of neuronal cells, whereas reduced
cell proliferation was recovered following treatment with midazolam
(Fig. 1A and B). In addition, in
neuronal cells, BSO and H2O2 activated
caspase-3 and caspase-9, followed by cleavage of PARP, and
activation of Bid and Bcl2, thus indicating activation of the
mitochondrial intrinsic apoptosis pathway (Fig. 1C). Exposure to BSO and
H2O2 increased the intracellular redox status
of cortical neuronal cells, as assessed by DCF fluorescence
intensity. Conversely, treatment of oxidative stress-induced
cortical neuronal cells with midazolam resulted in reduced DCF
intensity, and thus reduced levels of intracellular ROS (Fig. 2A and B). The ROS-lowering effects
of midazolam were further confirmed by co-treatment of cells with
midazolam and the known antioxidant agent, trolox. The combination
of trolox and midazolam more effectively reduced ROS levels and
elevated proliferation of neuronal cells, which was inhibited by
BSO and H2O2 (Fig. 2C and D). In addition, a previous
study demonstrated that midazolam exerts ROS-suppressing and cell
survival-modulating activity in neuroblastoma cells (30).
Oxidative stress-induced neuronal damage is
predominantly associated with activation of apoptotic cell death
pathways, with strong implications for JNK and ERK1/2 signaling
(31,32). ERK1/2 is an important member of the
mitogen-activated protein kinase (MAPK) family, which controls a
broad range of cellular activities and physiological processes.
ERK1/2 activation promotes cell survival and prevents against
oxidative stress. JNKs also belong to the MAPK family and they
regulate proliferation and differentiation functions in the cell.
Whether the JNK activation leads to cell proliferation or apoptosis
is dependent on the stimuli and the cell-type involved in such
activation (30,31). Previous reports have associated the
mitogen-activated protein kinase signaling cascades (ERK1/2 and
JNK) with oxidative stress-induced neuronal death (28,31,32);
similarly, the present study demonstrated that BSO and
H2O2 reduced protein levels of JNK, pJNK, ERK
and pERK in cortical neuronal cells (Fig. 3). Furthermore, midazolam was able
to activate JNK-ERK and NF-κB signaling pathways in oxidative
stress-induced cortical neuronal cells (Fig. 3). Activation of the NF-κB pathway
augments cell survival signaling via activation of ERK1/2-AKT
signaling, inactivation of proapoptotic proteins (Bid and
Bcl2-associated X protein) and upregulation of anti-apoptotic
proteins (Bcl2 and Bcl-extra large) (33). In addition, midazolam induced PKC-ε
activation in cortical neuronal cells, which correlates with the
suppression of ROS generation and apoptosis activation (Fig. 3). A previous study revealed that
BSO-induced ROS generation and neuronal cell death is
mechanistically mediated by suppression of PKC-ε, and trolox may
inhibit ROS elevation in BSO-treated cortical cells (21). Therefore, these results
demonstrated that midazolam may exert protective effects against
oxidative stress in neuronal cells via activation of JNK-ERK and
NF-κB signaling pathways, and elevation of PKC-ε, particularly in
BSO-treated neuronal cells.
In the present study, the effects of midazolam were
assessed in vivo in murine models of transient intraluminal
occlusion and ethanol-induced neuroapoptosis. MCAO is a model of
ischemic stroke and stress, and MCAO mice are characterized by
neuronal deficit, neuronal degeneration and cell death. Midazolam
post-conditioning resulted in improved neurological outcomes,
reduced neuronal deficits and infarct size, and reduced apoptosis
(Table I; Figs. 4 and 5). Midazolam infusion at the onset of
reperfusion decreased infarct volume and cell apoptosis, and
improved neurological deficits, thus indicating the protective
histological properties of midazolam. In addition, midazolam
reduced apoptotic TUNEL staining in MCAO mice, and reduced the
expression levels of caspase-3 and PARP. Exposure of ethanol to
neonatal mice during brain development can induce severe neuronal
apoptosis and degeneration (19).
Treatment of ethanol-treated mice with midazolam suppressed the
number of apoptotic cells in the brain sections (Fig. 6). Midazolam suppressed the elevated
levels of activated caspase-3 IHC staining and TUNEL staining in
the brain sections.
In conclusion, the present study emphasized the
effectiveness of midazolam against oxidative stress in neuronal
cells in vitro and in vivo. The results also
emphasized the clinical implications of midazolam as an anesthetic
that reduces pain during clinical and surgical procedures, which
also offers protection against neurodegenerative side effects.
Midazolam inhibited the generation of ROS and neuroapoptosis in
oxidative stress-induced cortical neuronal cells. Furthermore,
midazolam post-conditioning in MCAO mice improved neuronal
physiology, reduced neuronal deficits, and decreased infarct size
via suppression of ROS and inhibition of apoptosis. Midazolam also
suppressed apoptosis in ethanol-treated neonatal mice brains.
However, the dose and duration of anesthetic treatment remain
important considerations for neuronal cell growth and death. The
protective effects of midazolam are not solely dependent on
apoptotic suppression, and activation of the JNK-ERK cell survival
signaling pathway is additive to the effects of midazolam. The
results of the present study advocate the safe and protective
application of midazolam as a neuroprotective anesthetic agent.
References
|
1
|
So EC, Chang YT, Hsing CH, Poon PW, Leu SF
and Huang BM: The effect of midazolam on mouse Leydig cell
steroidogenesis and apoptosis. Toxicol Lett. 192:169–178. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jevtovic-Todorovic V, Hartman RE, Izumi Y,
Benshoff ND, Dikranian K, Zorumski CF, Olney JW and Wozniak DF:
Early exposure to common anesthetic agents causes widespread
neurodegeneration in the developing rat brain and persistent
learning deficits. J Neurosci. 23:876–882. 2003.PubMed/NCBI
|
|
3
|
Head BP, Patel HH, Niesman IR, Drummond
JC, Roth DM and Patel PM: Inhibition of p75 neurotrophin receptor
attenuates isoflurane-mediated neuronal apoptosis in the neonatal
central nervous system. Anesthesiology. 110:813–825. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vutskits L, Gascon E, Tassonyi E and Kiss
JZ: Clinically relevant concentrations of propofol but not
midazolam alter in vitro dendritic development of isolated
gamma-aminobutyric acid-positive interneurons. Anesthesiology.
102:970–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stratmann G, Sall JW, May LD, Bell JS,
Magnusson KR, Rau V, Visrodia KH, Alvi RS, Ku B, Lee MT and Dai R:
Isoflurane differentially affects neurogenesis and long-term
neurocognitive function in 60-day-old and 7-day-old rats.
Anesthesiology. 110:834–848. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Walker SM, Westin BD, Deumens R, Grafe M
and Yaksh TL: Effects of intrathecal ketamine in the neonatal rat:
Evaluation of apoptosis and long-term functional outcome.
Anesthesiology. 113:147–159. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Briner A, De Roo M, Dayer A, Muller D,
Habre W and Vutskits L: Volatile anesthetics rapidly increase
dendritic spine density in the rat medial prefrontal cortex during
synaptogenesis. Anesthesiology. 112:546–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Roo M, Klauser P, Briner A, Nikonenko
I, Mendez P, Dayer A, Kiss JZ, Muller D and Vutskits L: Anesthetics
rapidly promote synaptogenesis during a critical period of brain
development. PLoS One. 4:e70432009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yahalom B, Athiraman U, Soriano SG,
Zurakowski D, Carpino EA, Corfas G and Berde CB: Spinal anesthesia
in infant rats: Development of a model and assessment of neurologic
outcomes. Anesthesiology. 114:1325–1335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scallet AC, Schmued LC, Slikker W Jr,
Grunberg N, Faustino PJ, Davis H, Lester D, Pine PS, Sistare F and
Hanig JP: Developmental neurotoxicity of ketamine: Morphometric
confirmation, exposure parameters, and multiple fluorescent
labeling of apoptotic neurons. Toxicol Sci. 81:364–370. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abdel-Wahab AF and Al-Harizy WM: Propofol
protects against ischemia/reperfusion injury associated with
reduced apoptosis in rat liver. ISRN Anesthesiology 2013. Article
ID 517478. 82013.
|
|
12
|
Arai Y, Kondo T, Tanabe K, Zhao QL, Li FJ,
Ogawa R, Li M and Kasuya M: Enhancement of hyperthermia-induced
apoptosis by local anesthetics on human histiocytic lymphoma U937
cells. J Biol Chem. 277:18986–18993. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mishra SK, Kang JH, Lee CW, Oh SH, Ryu JS,
Bae YS and Kim HM: Midazolam induces cellular apoptosis in human
cancer cells and inhibits tumor growth in xenograft mice. Mol
Cells. 36:219–226. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Young C, Jevtovic-Todorovic V, Qin YQ,
Tenkova T, Wang H, Labruyere J and Olney JW: Potential of ketamine
and midazolam, individually or in combination, to induce apoptotic
neurodegeneration in the infant mouse brain. Br J Pharmacol.
146:189–197. 2005.PubMed/NCBI
|
|
15
|
Stevens MF, Werdehausen R, Gaza N,
Hermanns H, Kremer D, Bauer I, Küry P, Hollmann MW and Braun S:
Midazolam activates the intrinsic pathway of apoptosis independent
of benzodiazepine and death receptor signaling. Reg Anesth Pain
Med. 36:343–349. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Casellas P, Galiegue S and Basile AS:
Peripheral benzodiazepine receptors and mitochondrial function.
Neurochem Int. 40:475–486. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chiang T, Messing RO and Chou WH: Mouse
model of middle cerebral artery occlusion. J Vis Exp.
pii:27612011.
|
|
18
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Young C and Olney JW: Neuroapoptosis in
the infant mouse brain triggered by a transient small increase in
blood alcohol concentration. Neurobiol Dis. 22:548–554. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wüllner U, Seyfried J, Groscurth P,
Beinroth S, Winter S, Gleichmann M, Heneka M, Löschmann P, Schulz
JB, Weller M and Klockgether T: Glutathione depletion and neuronal
cell death: The role of reactive oxygen intermediates and
mitochondrial function. Brain Res. 826:53–62. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Valencia A and Morán J: Reactive oxygen
species induce different cell death mechanisms in cultured neurons.
Free Radic Biol Med. 36:1112–1125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Griffiths JD, Le NV, Grant S, Bjorksten A,
Hebbard P and Royse C: Symptomatic local anaesthetic toxicity and
plasma ropivacaine concentrations after transversus abdominis plane
block for Caesarean section. Br J Anaesth. 110:996–1000. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sakura S, Kirihara Y, Muguruma T,
Kishimoto T and Saito Y: The comparative neurotoxicity of
intrathecal lidocaine and bupivacaine in rats. Anesth Analg.
101:541–547. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang DX, Ding HZ, Jiang S, Zeng YM and
Tang QF: An in vitro study of the neuroprotective effect of
propofol on hypoxic hippocampal slice. Brain Inj. 28:1758–1765.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liang C, Cang J, Wang H and Xue Z:
Propofol attenuates cerebral ischemia/reperfusion injury partially
using heme oxygenase-1. J Neurosurg Anesthesiol. 25:311–316. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shao H, Zhang Y, Dong Y, Yu B, Xia W and
Xie Z: Chronic treatment with anesthetic propofol improves
cognitive function and attenuates caspase activation in both aged
and Alzheimer's disease transgenic mice. J Alzheimers Dis.
41:499–513. 2014.PubMed/NCBI
|
|
28
|
Shi C, Zhao L, Zhu B, Li Q, Yew DT, Yao Z
and Xu J: Protective effects of Ginkgo biloba extract (EGb761) and
its constituents quercetin and ginkgolide B against beta-amyloid
peptide-induced toxicity in SH-SY5Y cells. Chem Biol Interact.
181:115–123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bournival J, Quessy P and Martinoli MG:
Protective effects of resveratrol and quercetin against MPP+
-induced oxidative stress act by modulating markers of apoptotic
death in dopaminergic neurons. Cell Mol Neurobiol. 29:1169–1180.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chong WS, Hyun CL, Park MK, Park JM, Song
HO, Park T, Lim YS, Cho CK, Kang PS and Kwon HU: Midazolam protects
B35 neuroblastoma cells through Akt-phosphorylation in reactive
oxygen species derived cellular injury. Korean J Anesthesiol.
62:166–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Satoh T, Nakatsuka D, Watanabe Y, Nagata
I, Kikuchi H and Namura S: Neuroprotection by MAPK/ERK kinase
inhibition with U0126 against oxidative stress in a mouse neuronal
cell line and rat primary cultured cortical neurons. Neurosci Lett.
288:163–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW
and DeFranco DB: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|