Introduction
Osteoarthritis (OA) is a common degenerative
disorder of joints characterized by the progressive breakdown of
articular cartilage. It is a global public health problem that
causes substantial pain and disability in middle-aged and older
individuals, and leads to an economic burden and a reduced quality
of life (1,2). However, no curative therapeutics are
currently available for OA. Currently, the standard treatment for
OA focuses on the control of pain and improvement of joint
function. The most commonly prescribed medications include locally
administered corticosteroids (e.g., prednisone and betamethasone),
nonsteroidal anti-inflammatory drugs (NSAIDs; e.g., piroxicam,
ibuprofen and indomethacin) and other anti-inflammatory drugs. It
is well established that the development and progression of OA are
associated with inflammation even in the early phase of the disease
(3,4). Inflammatory molecules, including
pro-inflammatory cytokines, are reported to be important mediators
involved in the pathophysiology of OA (5). Tumor necrosis factor (TNF)-α and
interleukin (IL)-1β are the major pro-inflammatory cytokines that
lead to significant breakdown of the cartilage macromolecules
(5,6). They act independently or in
conjunction with other cytokines to initiate inflammation. Previous
studies have demonstrated that TNF-α and IL-1β directly
downregulate the expression of extracellular matrix components,
including aggrecan and type II collagen (7,8) and
increase the expression of catabolic factors, including matrix
metalloproteinase (MMP)-1 and MMP-13 (9,10).
IL-10 protects cartilage, and stimulates the expression of type II
collagen and proteoglycan (11,12).
It reverses the damaging effects of TNF-α and IL-1β on cartilage by
inhibiting the production of MMPs, pro-inflammatory cytokines and
nitric oxide (13). IL-6 is known
to have pro-inflammatory and anti-inflammatory effects. IL-6
release is stimulated by TNF-α and it is thought to upregulate the
number of inflammatory cells in synovial tissue (14). However, other studies have
suggested that IL-6 negatively regulates further expression of
TNF-α (14). In addition to the
aforementioned pro-inflammatory cytokines, chemokines are also
implicated in OA (5). IL-8 is a
chemokine that is synthesized in OA chondrocytes and has an
important role in the pathogenesis of OA. It promotes the
production of MMP-1 and MMP-13 by articular chondrocytes (15). However, limited information is
available regarding studies on the therapeutic mechanisms of
corticosteroids and NSAIDs in OA. Therefore, the aim of the current
study was to identify the potential underlying mechanisms. The
present study employed prednisone, betamethasone, piroxicam,
ibuprofen and indomethacin to investigate therapeutic mechanisms in
OA. The findings of the present study may help to guide the
clinical management of OA.
Materials and methods
Cell culture
CHON-002 human chondrocyte cell line, derived from
the long bone of an 18-week-old female fetus, was purchased from
the American Type Culture Collection (Manassas, VA, USA). The
CHON-002 cell line was cultured in Dulbecco's modified Eagle's
medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 0.1 mg/ml G-418, 10% fetal bovine serum (FBS;
Thermo Fisher Scientific, Inc.), 100 U/ml penicillin (Thermo Fisher
Scientific, Inc.) and 100 mg/ml streptomycin (Thermo Fisher
Scientific, Inc.) in a humidified atmosphere of 5% CO2
at 37°C.
Drug preparation and treatment
All drugs (piroxicam, ibuprofen, indomethacin,
prednisone and betamethasone) were purchased from Sigma-Aldrich;
Merck Millipore (Darmstadt, Germany) as powders. Drugs were
prepared as the following treatment groups: Piroxicam (5 µM),
ibuprofen (400 µM), indomethacin (100 µM), prednisone (5 µM) and
betamethasone (10 µM). Dimethyl sulfoxide (10 µl) was applied to
fully dissolve 1 mg of each drug. Subsequently, the dissolved
mixture was diluted with serum-free F12K media (Sigma-Aldrich;
Merck Millipore) supplemented with 5 ml penicillin, 5 ml
streptomycin and 50 µg/ml ascorbic acid. Additionally, all
cytokines (TNF-α, IL-1β, IL-6, IL-8 and IL-10) were obtained in
lyophilized powdered form from R&D Systems, Inc. (Minneapolis,
MN, USA) and dissolved in sterile 4 mM hydrochloric acid
supplemented with 1 mg/ml bovine serum albumin. Cytokines were
prepared as the following treatment groups: TNF-α (5 ng/ml), IL-1β
(1 ng/ml), IL-6 (10 ng/ml), IL-8 (10 ng/ml) and IL-10 (5 ng/ml). In
addition, an inhibitor of the proteasome, MG132 (10 µM; Selleck
Chemicals, Inc., Houston, TX, USA) was added to inhibit NF-κB by
preventing the degradation of the inhibitory subunit of NF-κB
(IκBα).
Cell viability assay
Cells were plated onto 96-well plates at a final
concentration of 1×104 cells/well in culture medium with drugs
(piroxicam, ibuprofen, indomethacin, prednisone and betamethasone)
or cytokines (TNF-α, IL-1β, IL-6, IL-8 and IL-10). Untreated cells
were considered as controls. At 48 h after treatment, 5 mg/ml MTT
(20 µl; Sigma-Aldrich; Merck Millipore) was added to each well,
followed by incubation for 4 h at 37°C. Thereafter,
dimethylsulfoxide (DMSO; Sigma-Aldrich; Merck Millipore) was added
to dissolve the formazan. Absorbance at 570 nm was measured using a
Synergy plate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
ELISA
After 48 h of treatment, the conditioned medium was
collected for ELISA. Levels of IL-1β, IL-6, IL-8 and IL-10 in cells
with or without TNF-α treatment, and levels of IL-6 and IL-8 in
cells with or without TNF-α treatment, drugs (piroxicam, ibuprofen,
indomethacin, prednisone and betamethasone) or TNF-α in combination
with drugs were respectively determined by human IL-1β ELISA Kit
(cat. no. CSB-E08053h), human IL-6 ELISA Kit (cat. no.
CSB-E04638h), human IL-8 ELISA Kit (cat. no. CSB-E04641h) and human
IL-10 ELISA Kit (cat. no. CSB-E04593h) according to the
manufacturer's protocol. All ELISA kits were purchased from
Flarebio Biotech LLC (College Park, MD, USA).
Small interfering RNA (siRNA)
transfection
Cells in the exponential growth phase were seeded in
a 60 mm dish at a concentration of 5×105 cells/well. Transfection
was performed when cells reached 70% confluence. Signal transducer
and activator of transcription (STAT) 3 was silenced through the
use of an siRNA targeting STAT3 (siSTAT3). siSTAT3 and negative
control siRNA-were designed and constructed by Shanghai GenePharma
Co., Ltd. (Shanghai, China). The targeting sequence for siSTAT3 was
5′-CAGGCTGGTAATTTATATAAT-3′ and negative control siRNA was
5′-CATTGACTTATAAATTCGTTC-3′. After 24 h of incubation, cells were
transfected using Lipofectamine® 2000 (reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. At 48 h after the transfection, the cells were
prepared for western blot analysis.
Protein isolation and western blot
analysis
Following stimulation with or without cytokines or
drugs, total cellular protein and nuclear protein were extracted
from the cells using nuclear and cytoplasmic extraction reagent
kits (Beyotime Institute of Biotechnology, Inc., Haimen, China) and
stored at −80°C until use. Protein levels of STAT3, phosphorylated
STAT3 (pSTAT3Ser727 and pSTAT3Tyr705), collagen I, MMP-1, MMP-13,
aggrecan and IκB were assessed using total cellular protein, while
protein levels of NF-κB subunit p65 were assessed using total
cellular protein and nuclear protein. The protein concentration was
quantified using a bicinchoninic acid protein assay kit (Pierce;
Thermo Fisher Scientific, Inc.). Protein samples (20 µg per lane)
were resolved on a 10–12% SDS-PAGE gel and blotted onto PVDF or
nitrocellulose membranes, blocked in 5% fresh non-fat milk in
phosphate buffered saline for 2 h at room temperature and probed
with primary antibodies against pSTAT3Ser727 (cat. no. 9134;
1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA),
pSTAT3Tyr705 (cat. no. 4093; 1:1,000; Cell Signaling Technology,
Inc.), STAT3 (cat. no. 12640; 1:1,000; Cell Signaling Technology,
Inc.), collagen I (cat. no. sc-29009; 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), MMP-1 (cat. no. sc-137044;
1:1,000; Santa Cruz Biotechnology, Inc.), MMP-13 (cat. no.
sc-12363; 1:1,000; Santa Cruz Biotechnology, Inc.), aggrecan (cat.
no. sc-70332; 1:1,000; Santa Cruz Biotechnology, Inc.), IκB (cat.
no. BS2224; 1:1,000; Bioworld Technology, Inc., St. Louis Park, MN,
USA), and p65 (cat. no. 8242; 1:1,000; Cell Signaling Technology,
Inc.) overnight at 4°C. Lamin B (cat. no. 12255; 1:1,000; Cell
Signaling Technology, Inc.) was used as an internal nuclear protein
loading control, while GAPDH (cat. no. 5174; 1:1,000; Cell
Signaling Technology, Inc.) was used as an internal total protein
loading control. The membranes were then incubated with secondary
IgG conjugated to horseradish peroxidase (cat. no. 7071; 1:5,000;
Cell Signaling Technology, Inc.) for 1 h at room temperature.
Immunoreactive protein bands were visualized by enhanced
chemiluminescence western blotting substrate (Pierce; Thermo Fisher
Scientific, Inc.). The immunoreactive bands were analyzed using
Image Gauge version 4.0 software (Fujifilm, Tokyo, Japan). Each
condition was carried out in triplicate.
Statistical analysis
Data are expressed as the mean ± standard deviation.
SPSS version no. 17.0 software (SPSS, Inc., Chicago, IL, USA) was
used for statistical analysis. Student's t-test was performed to
calculate P-values for 2 groups, or one-way analysis of variance
followed by Tukey-Kramer's post hoc test for multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Expression levels of IL-1β, IL-6, IL-8
and IL-10 with or without TNF-α treatment
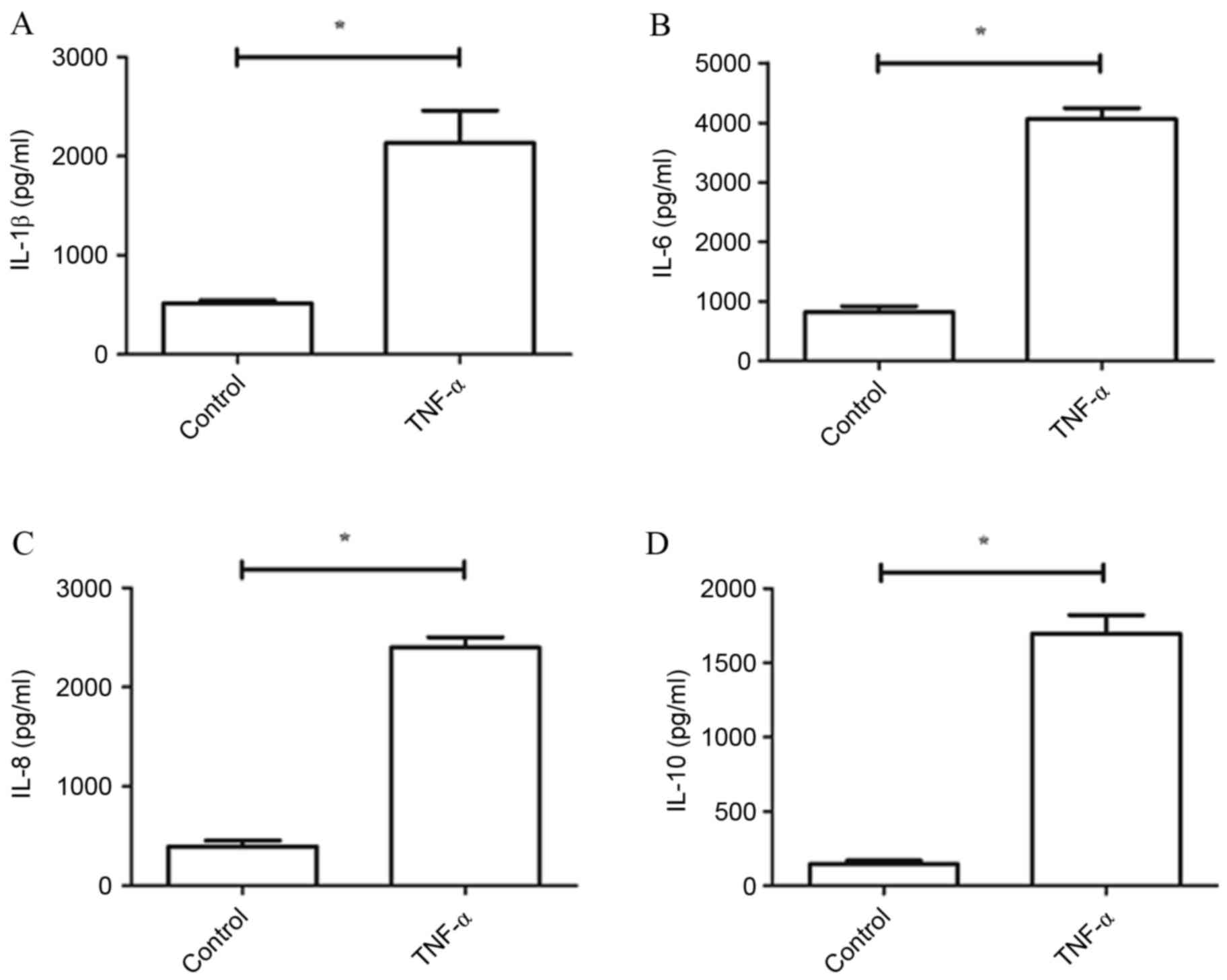
The present study used doses of TNF-α (5 ng/ml) to
activate cultured chondrocytes and to stimulate an inflammatory
response in vitro. Primary cultures that were not treated
with TNF-α served as a control group. Levels of IL-1β, IL-6, IL-8
and IL-10 were determined by ELISA. Compared with the control
group, expression levels of IL-1β, IL-6, IL-8 and IL-10 were all
significantly increased by TNF-α (P<0.05; Fig. 1).
Expression of collagen I, aggrecan,
MMP-1 and MMP-13 with or without cytokines
Western blotting was performed to observe the
effects of cytokines, including IL-1β, IL-6, IL-8 and IL-10, on the
protein expression of collagen I, aggrecan, MMP-1 and MMP-13. The
results demonstrated that expression levels of collagen I, MMP-1
and MMP-13 were all markedly increased by treatment with IL-6 or
IL-8. However, there were no observable differences in protein
levels of aggrecan when treated with IL-1β, IL-6, IL-8 or IL-10
compared with the control group. Although expression levels of
collagen I, MMP-1 and MMP-13 were elevated by treatment with IL-1β
or IL-10, there was no statistically significant difference between
the experimental and the control groups (Fig. 2A).
 | Figure 2.Effects of cytokines on expression of
collagen I, aggrecan, MMP-1, MMP-13, STAT3 and NF-κB p65 signaling
pathways. (A) Effects of IL-1β, IL-6, IL-8 and IL-10 on expression
of collagen I, aggrecan, MMP-1 and MMP-13. (B) Effects of IL-6 and
IL-8 expression on STAT3 and NF-κB p65 signaling pathways. IL,
interleukin; MMP, matrix metalloproteinase; STAT, signal transducer
and activator of transcription; NF-κB, nuclear factor-κB; IκB,
inhibitory subunit of NF-κB; pSTAT3, phosphorylated STAT3. |
Promotion of STAT3 phosphorylation and
activation of NF-κB subunit p65 by IL-6 and IL-8
To investigate the associations between the STAT3 or
NF-κB p65 signaling pathways and IL-6 and IL-8, the present study
determined the levels of pSTAT3Ser727, pSTAT3Tyr705, STAT3, IκB and
p65 following stimulation with IL-6 or IL-8. As presented in
Fig. 2B, the expression levels of
pSTAT3Ser727 and pSTAT3Tyr705 were visibly increased by treatment
with IL-6 or IL-8 compared with the control group, indicating that
STAT3 phosphorylation was promoted by IL-6 and IL-8. Additionally,
following treatment with IL-6 or IL-8, the expression of IκB was
lower than the control group, while the expression of p65 (both in
the cell nucleus and whole cell) was higher than the control group,
suggesting that NF-κB p65 was activated by stimulation by IL-6 or
IL-8.
Effects of STAT3 and NF-κB inhibition
on expression of collagen I, aggrecan, MMP-1 and MMP-13
To investigate whether IL-6 and IL-8 affect the
expression of collagen I, MMP-1, and MMP-13 via STAT3 and NF-κB p65
signaling pathways, the expression of STAT3 was silenced and NF-κB
was inhibited by use of MG132. As shown in Fig. 3A, the expression of pSTAT3Ser727
and pSTAT3Tyr705 were visibly decreased following transfection with
siSTAT3 compared with the control. Following siSTAT3 transfection,
the effects of IL-6 and IL-8 on the expression of collagen I, MMP-1
and MMP-13 were decreased, but the effects were not completely
eliminated, indicating that other mechanisms exist for the
regulation of collagen I, MMP-1 and MMP-13 expression. However,
expression of collagen I, MMP-1 and MMP-13 was reduced when treated
with the MG132 and transfected with siSTAT3 compared with the
IL-6/IL-8+ siSTAT3 group (Fig. 3B and
C). The results demonstrated that the effects of IL-6 and IL-8
on the expression of collagen I, MMP-1, and MMP-13 may be through
promotion of STAT3 phosphorylation and activation of NF-κB p65.
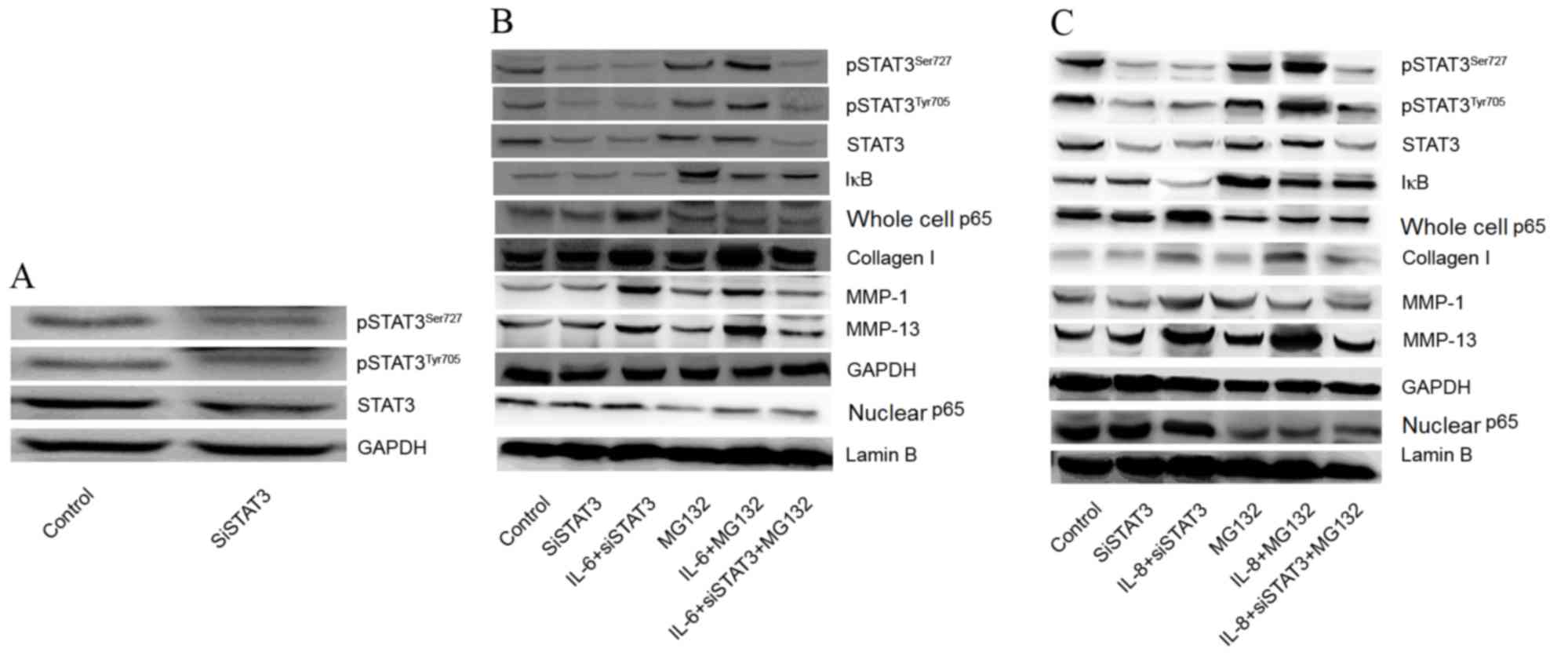 | Figure 3.Effects of STAT3 and NF-κB inhibition
on expression of collagen I, aggrecan, MMP-1 and MMP-13. (A)
Effects of siSTAT3 on expression of STAT3. (B) Effects of IL-6
combined with STAT3 and NF-κB inhibition (using MG132) on the
expression of collagen I, aggrecan, MMP-1 and MMP-13. (C) Effects
of IL-8 combined with STAT3 and NF-κB inhibition on expression of
collagen I, aggrecan, MMP-1 and MMP-13. IL, interleukin; MMP,
matrix metalloproteinase; STAT, signal transducer and activator of
transcription; NF-κB, nuclear factor-κB; IκB, inhibitory subunit of
NF-κB; siSTAT3, small interfering RNA-STAT3; pSTAT3, phosphorylated
STAT3. |
Effects of drugs and cytokines on cell
viability and expression of IL-6 and IL-8
Compared with the control group, no change in cell
viability was observed following treatment with cytokines (TNF-α,
IL-1β, IL-6, IL-8 and IL-10) or drugs (piroxicam, ibuprofen,
indomethacin, prednisone and betamethasone). There were no
statistically significant differences in cell viability between
groups, and the average cell viability was >90% (Fig. 4A and B). Additionally, no
significant differences were observed in the expression of IL-6 and
IL-8 measured by ELISA following treatment with drugs alone.
However, the increased levels of IL-6 and IL-8 induced by TNF-α
were significantly decreased when combined with administration of
prednisone, ibuprofen or betamethasone (P<0.05), whereas no
significant differences were found between the levels of IL-6 and
IL-8 following stimulation by TNF-α alone compared with TNF-α
combined with administration of piroxicam or indomethacin (Fig. 4C and D).
Effect of IL-6 or IL-8 combined with
drugs on STAT3, NF-kB p65, collagen I, MMP-1 and MMP-13
expression
To investigate the effects of drugs on expression of
STAT3 and NF-kB p65 signaling pathways, as well as the production
of Collagen I, MMP-1 and MMP-13, we exposed the cells to drug and
IL-6 or IL-8 combinations. The present study observed that the
expression of pSTAT3Ser727, pSTAT3Tyr705, p65 (both in the cell
nucleus and whole-cell), collagen I, MMP-1 and MMP-13 were reduced,
while the levels of IκB were increased by administration of
prednisone, ibuprofen and betamethasone. These results suggested
that prednisone, ibuprofen and betamethasone decreased the
inflammatory response and prevented abnormal catabolism (Fig. 5A and B).
 | Figure 5.Effects of IL-6-drug and IL-8-drug
combinations on STAT3, NF-κB p65, collagen I, MMP-1 and MMP-13
expression. (A) Effect of IL-6-drug combinations on STAT3, NF-κB
p65, collagen I, MMP-1 and MMP-13 expression. (B) Effect of IL-8
plus drugs on STAT3, NF-κB p65, collagen I, MMP-1 and MMP-13
expression. IL, interleukin; MMP, matrix metalloproteinase; STAT,
signal transducer and activator of transcription; NF-κB, nuclear
factor-kappa B; IκB, inhibitory subunit of NF-κB; pSTAT3,
phosphorylated STAT3. |
Discussion
The present study demonstrated that cultured
chondrocytes are activated by TNF-α, which leads to increased
expression of IL-1β, IL-6, IL-8 and IL-10, which are associated
with OA. Of the cytokines investigated in the present study, IL-6
and IL-8 increased the levels of collagen I, MMP-1 and MMP-13,
promoted STAT3 phosphorylation and activated NF-κB p65.
Additionally, corticosteroids (prednisone and betamethasone) and
NSAIDs (ibuprofen) reduced the expression of IL-6 and IL-8,
inactivated STAT3 and NF-κB signaling pathways, and reduced the
expression of collagen I, MMP-1 and MMP-13, which protect against
OA. In order to observe the efficacy of NSAIDs and steroids in
preventing OA, TNF-α, at an optimized concentration of 5 ng/ml, was
used to stimulate the cultured chondrocytes and to mimic a
catabolic environment in vitro in the present study
(16,17). The role of pro-inflammatory
mediators in the pathophysiology of OA has been extensively
investigated (4,17,18).
An improved understanding of the role of cytokines involved in the
pathophysiology of OA is important for the identification of
potential therapeutic targets. Pro-inflammatory cytokines and
chemokines locally produced by chondrocytes contribute to the
pathological processes in OA. TNF-α, IL-1β, IL-6, IL-8 and IL-10
enhance inflammation of the joints and subsequently lead to the
degradation of cartilage (5). It
has been reported that TNF-α may induce the production of IL-6
(19), IL-8 (20) and IL-10 (21). Corresponding with previous studies,
the present study also observed that chondrocytes were activated by
TNF-α. Expression levels of IL-6, IL-8 and IL-10 were all
significantly increased by TNF-α addition. In addition, the levels
of IL-1β were also increased by TNF-α. Elevated IL-6 and IL-8
levels have been previously described in the serum and synovial
fluid of patients with OA (22,23).
When combined with IL-1β, the expression of type II collagen was
suppressed and the release of MMP-1 and MMP-13 was stimulated by
IL-6 (24,25). Furthermore, the production of
tissue inhibitors of metalloproteinases can be induced by IL-6,
thereby, suppressing proteolytic damage induced by MMPs (14). Additionally, animal experimental
studies have suggested that IL-6-deficient mice can spontaneously
develop OA and that the cartilage repair response is damaged in
these mice (26). IL-8, also known
to be an osteoclastogenic factor, is responsible for
osteoclastogenesis and the bone resorption process, which is
involved in the OA cartilage degradation process (27). Furthermore, IL-8 promotes the
activation of mononuclear cell leukocytes into the synovium,
degranulation of neutrophils, production of MMP-13 by chondrocytes,
apoptosis of chondrocytes and loss of proteoglycans (28–30).
The present study observed that IL-6 and IL-8 elevated the levels
of collagen I, MMP-1 and MMP-13, and also increased STAT3
phosphorylation and NF-κB p65 levels. Previous studies have
demonstrated that NF-κB (p65/p50), NF-κB/IκB and STAT3 signaling
are abnormally activated in OA chondrocytes (4,31,32).
The NF-κB pathway is an essential regulator of the inflammatory
cytokine-induced catabolic response in chondrocytes. Activation of
NF-κB (p65/p50) signaling is vital to allow chondrocytes to induce
production of MMPs, nitric oxide synthase 2, cyclooxygenase-2 and
IL-1 (4). NF-κB mediates the
expression of pro-inflammatory cytokines including TNF-α, IL-1β and
IL-6, and chemokines (33).
Latourte et al (34)
suggested that IL-6 has various catabolic effects on cartilage,
predominantly regulated by STAT3. Blockade of STAT3 protected
against OA induced by destabilization of the medial meniscus in
mice (34). Therefore, it was
speculated that the effects of IL-6 and IL-8 on collagen I, MMP-1
and MMP-13 may be via STAT3 and NF-κB p65 signaling pathways. To
confirm the hypothesis, the present study silenced the expression
of STAT3 and inhibited NF-κB, using the proteasome inhibitor MG132,
and then incubated chondrocytes with IL-6 or IL-8. When combined
with siSTAT3 and MG132, IL-6 and IL-8 significantly reduced the
levels of collagen I, MMP-1 and MMP-13, which confirmed the
hypothesis. To further investigate the therapeutic mechanisms of
NSAIDs and steroids in OA, piroxicam, ibuprofen, indomethacin,
prednisone and betamethasone were used. No change in cell viability
following treatment with these five drugs or cytokines was observed
in chondrocytes compared with cells receiving no treatment. In
combination with TNF-α, prednisone, betamethasone and ibuprofen
significantly reduced the expression of IL-6 and IL-8.
Additionally, chondrocytes were co-cultured with drugs and IL-6 or
IL-8 and it was observed that prednisone, betamethasone and
ibuprofen reduced the phosphorylation of STAT3 and activation of
NF-κB, and also inhibited the expression of collagen I, MMP-1 and
MMP-13. Thus, these three drugs exert a variety of protective
effects in OA.
In conclusion, the results of the present study
suggest that the ability of ibuprofen, prednisone and betamethasone
to protect against OA results from inhibition of IL-6 and IL-8
expression, subsequent suppression of NF-κB and STAT3 signaling
pathways, and, ultimately, reduction of collagen I, MMP-1 and
MMP-13 expression. The results of the present study might provide
an experimental basis for the treatment of OA and guide the
clinical use of drugs including ibuprofen, prednisone and
betamethasone.
References
|
1
|
Cross M, Smith E, Hoy D, Nolte S, Ackerman
I, Fransen M, Bridgett L, Williams S, Guillemin F, Hill CL, et al:
The global burden of hip and knee osteoarthritis: Estimates from
the global burden of disease 2010 study. Ann Rheum Dis.
73:1323–1330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hunter DJ, Schofield D and Callander E:
The individual and socioeconomic impact of osteoarthritis. Nat Rev
Rheumatol. 10:437–441. 2014.PubMed/NCBI
|
|
3
|
Felson DT: Clinical practice.
Osteoarthritis of the knee. N Engl J Med. 354:841–848. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Goldring MB and Otero M: Inflammation in
osteoarthritis. Curr Opin Rheumatol. 23:471–478. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapoor M, Martel-Pelletier J, Lajeunesse
D, Pelletier JP and Fahmi H: Role of proinflammatory cytokines in
the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 7:33–42.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martel-Pelletier J: Pathophysiology of
osteoarthritis. Osteoarthritis Cartilage. 12 Suppl A:S31–S33. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saklatvala J: Tumour necrosis factor alpha
stimulates resorption and inhibits synthesis of proteoglycan in
cartilage. Nature. 322:547–549. 1986. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Goldring MB, Fukuo K, Birkhead JR, Dudek E
and Sandell LJ: Transcriptional suppression by interleukin-1 and
interferon-gamma of type II collagen gene expression in human
chondrocytes. J Cell Biochem. 54:85–99. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richardson DW and Dodge GR: Effects of
interleukin-1beta and tumor necrosis factor-alpha on expression of
matrix-related genes by cultured equine articular chondrocytes. Am
J Vet Res. 61:624–630. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cruz R, Miranda-Sánchez M, Solís-García DB
and Kouri J: Recent patents on metalloproteinases as biomarkers in
osteoarthritis diagnosis and treatment. Recent Patents on
Biomarkers. 4:1–10. 2014. View Article : Google Scholar
|
|
11
|
Müller RD, John T, Kohl B, Oberholzer A,
Gust T, Hostmann A, Hellmuth M, Laface D, Hutchins B, Laube G, et
al: IL-10 overexpression differentially affects cartilage matrix
gene expression in response to TNF-alpha in human articular
chondrocytes in vitro. Cytokine. 44:377–385. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jansen NW, Roosendaal G, Hooiveld MJ,
Bijlsma JW, van Roon JA, Theobald M and Lafeber FP: Interleukin-10
protects against blood-induced joint damage. Br J Haematol.
142:953–961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Y and Lou S: Direct protective effect
of interleukin-10 on articular chondrocytes in vitro. Chin Med J
(Engl). 114:723–725. 2001.PubMed/NCBI
|
|
14
|
Fernandes JC, Martel-Pelletier J and
Pelletier JP: The role of cytokines in osteoarthritis
pathophysiology. Biorheology. 39:237–246. 2002.PubMed/NCBI
|
|
15
|
Borzi RM, Mazzetti I, Marcu KB and
Facchini A: Chemokines in cartilage degradation. Clin Orthop Relat
Res. 427:S53–S61. 2004. View Article : Google Scholar
|
|
16
|
Cho H, Lee S, Park SH, Huang J, Hasty KA
and Kim SJ: Synergistic effect of combined growth factors in
porcine intervertebral disc degeneration. Connect Tissue Res.
54:181–186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cho H, Walker A, Williams J and Hasty KA:
Study of osteoarthritis treatment with anti-inflammatory drugs:
Cyclooxygenase-2 inhibitor and steroids. Biomed Res Int.
2015:5952732015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sokolove J and Lepus CM: Role of
inflammation in the pathogenesis of osteoarthritis: Latest findings
and interpretations. Ther Adv Musculoskelet Dis. 5:77–94. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guerne PA, Carson DA and Lotz M: IL-6
production by human articular chondrocytes. Modulation of its
synthesis by cytokines, growth factors, and hormones in vitro. J
Immunol. 144:499–505. 1990.PubMed/NCBI
|
|
20
|
Lotz M, Terkeltaub R and Villiger PM:
Cartilage and joint inflammation. Regulation of IL-8 expression by
human articular chondrocytes. J Immunol. 148:466–473.
1992.PubMed/NCBI
|
|
21
|
Mrosewski I, Jork N, Gorte K, Conrad C,
Wiegand E, Kohl B, Ertel W, John T, Oberholzer A, Kaps C and
Schulze-Tanzil G: Regulation of osteoarthritis-associated key
mediators by TNFα and IL-10: Effects of IL-10 overexpression in
human synovial fibroblasts and a synovial cell line. Cell Tissue
Res. 357:207–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaneko S, Satoh T, Chiba J, Ju C, Inoue K
and Kagawa J: Interleukin-6 and interleukin-8 levels in serum and
synovial fluid of patients with osteoarthritis. Cytokines Cell Mol
Ther. 6:71–79. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Valcamonica E, Chighizola CB, Comi D, De
Lucia O, Pisoni L, Murgo A, Salvi V, Sozzani S and Meroni PL:
Levels of chemerin and interleukin 8 in the synovial fluid of
patients with inflammatory arthritides and osteoarthritis. Clin Exp
Rheumatol. 32:243–250. 2014.PubMed/NCBI
|
|
24
|
Rowan AD, Koshy PJ, Shingleton WD, Degnan
BA, Heath JK, Vernallis AB, Spaull JR, Life PF, Hudson K and
Cawston TE: Synergistic effects of glycoprotein 130 binding
cytokines in combination with interleukin-1 on cartilage collagen
breakdown. Arthritis Rheum. 44:1620–1632. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Poree B, Kypriotou M, Chadjichristos C,
Beauchef G, Renard E, Legendre F, Melin M, Gueret S, Hartmann DJ,
Malléin-Gerin F, et al: Interleukin-6 (IL-6) and/or soluble IL-6
receptor down-regulation of human type II collagen gene expression
in articular chondrocytes requires a decrease of Sp1.Sp3 ratio and
of the binding activity of both factors to the COL2A1 promoter. J
Biol Chem. 283:4850–4865. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Malemud CJ: Anticytokine therapy for
osteoarthritis: Evidence to date. Drugs Aging. 27:95–115. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goldring MB: The role of cytokines as
inflammatory mediators in osteoarthritis: Lessons from animal
models. Connect Tissue Res. 40:1–11. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Matsukawa A, Yoshimura T, Maeda T,
Ohkawara S, Takagi K and Yoshinaga M: Neutrophil accumulation and
activation by homologous IL-8 in rabbits. IL-8 induces destruction
of cartilage and production of IL-1 and IL-1 receptor antagonist in
vivo. J Immunol. 154:5418–5425. 1995.PubMed/NCBI
|
|
29
|
Olson TS and Ley K: Chemokines and
chemokine receptors in leukocyte trafficking. Am J Physiol Regul
Integr Comp Physiol. 283:R7–R28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Borzi RM, Mazzetti I, Magagnoli G,
Paoletti S, Uguccioni M, Gatti R, Orlandini G, Cattini L and
Facchini A: Growth-related oncogene alpha induction of apoptosis in
osteoarthritis chondrocytes. Arthritis Rheum. 46:3201–3211. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olivotto E, Otero M, Marcu KB and Goldring
MB: Pathophysiology of osteoarthritis: Canonical
NF-κB/IKKβ-dependent and kinase-independent effects of IKKα in
cartilage degradation and chondrocyte differentiation. RMD Open. 1
Suppl 1:e0000612015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayashi S, Nishiyama T, Hashimoto S,
Fujishiro T, Kanzaki N, Iwasa K, Sakata S, Chinzei N, Kuroda R and
Kurosaka M: P21 regulates MMP-13 expression through STAT3 signaling
in chondrocytes. Osteoarthritis and Cartilage. 21 Suppl:S46–S47.
2013. View Article : Google Scholar
|
|
33
|
Sultana F and Rasool M: A novel
therapeutic approach targeting rheumatoid arthritis by combined
administration of morin, a dietary flavanol and non-steroidal
anti-inflammatory drug indomethacin with reference to
pro-inflammatory cytokines, inflammatory enzymes, RANKL and
transcription factors. Chem Biol Interact. 230:58–70. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Latourte A, Cherifi C, Korng EH, Bouaziz
W, Brentano TF, Cohen-Solal M, Haÿ E and Richette P: Inhibition of
the interleukin-6-induced STAT3 signalling pathway is
chondroprotective. ECTS-IBMS. 2015.
|